Präsentation herunterladen
Die Präsentation wird geladen. Bitte warten

1
9. Winterschool Obergurgl, 10. bis 14
9. Winterschool Obergurgl, 10. bis 14. März 2013 CF und andere seltene Lungenerkrankungen a1 – Proteaseinhibitormangel alpha Antitrypsinmangel Wolfgang Gleiber Medizinische Klinik 1 Schwerpunkt Pneumologie/Allergologie Johann Wolfgang Goethe-Universität Frankfurt am Main

2
Einführung Der Alpha-1-Antitrypsinmangel beruht auf einer genetisch bedingten Störung, die sich klinisch als chronische Bronchitis, Lungenemphysem, Leber- und sehr selten als Hauterkrankung manifestiert. Das gleichzeitige Auftreten von Lungen- und Leberschäden im selben Individuum kommt nur sehr selten vor. Pannikulitis, entzündliche Veränderung des Fettgewebes. Oft ausgedehnte Nekrosen 2

6
Panlobulär – Diffuser Befall, im Lobulus werden alle Alveolen zerstört
Panlobulär – Diffuser Befall, im Lobulus werden alle Alveolen zerstört. Vorzugsweise sind die basalen Lungenabschnitte betroffen. Zentrilobulär – von den Bronchioli respiratorii ausgehend, vorzugsweise in den apikalen Abschnitten 6

12
Häufigkeit Anfang der 60er Jahre Erstbeschreibung 1:1500 – 1:5000
Ca bis homozygote Merkmalsträger in Deutschland Hauptsächlich weiße Bevölkerung europäischer Abstammung Diagnosestellung im Schnitt 5,7 Jahre nach Beginn der pulmonalen Symptomatik Durchschnittsalter bei Diagnose 41 Jahre Mittlere Lebenserwartung von homozygoten Mangelträgern bei Rauchern ca. 50 Jahre Mittlere Lebenserwartung von homozygoten Mangelträgern bei Nichtrauchern ca. 65 Jahre Die Schweden Carl-Bertil Laurell und Sten Eriksson beschrieben die Erkrankung als erste in den frühen 60er Jahren. In einer Familie waren Emphyseme gehäuft aufgetreten. In Deutschland beträgt die Zahl der homozygot Betroffenen etwa , davon ist schätzungsweise ein Drittel manifest erkrankt. Der Großteil aller Alpha-1-Antitrypsin-Mangelträger ist heute nicht identifiziert oder wird unter anderen Diagnosen, wie zum Beispiel Asthma bronchiale oder chronische obstruktive Lungenerkrankung (COPD) geführt In Deutschland wird die Prävalenz mit 8000 bis homozygot Merkmalsträgern eines Alpha-1-PI-Man- 12
 geführt. In Deutschland wird die Prävalenz mit 8000 bis homozygot Merkmalsträgern eines Alpha-1-PI-Man- 12.")
13
Alpha-1-Antitrypsin-Mangel autosomal rezessiv
Das Alpha-1 Antitrypsin-Gen befindet sich auf Chromosom 14 am Gen-Ort q32.1 Mutationen im genetischen Code bewirken eine verminderte oder fehlerhafte Synthese und Freisetzung von Alpha-1 Antitrypsin Signifikant verminderte Serumkonzentration wenn beide Allele (von beiden Elternteilen) abnormale Sequenzen aufweisen, also ein homozygoter Mangel vorliegt. Normalerweise beträgt die tägliche Syntheserate circa 34 mg/kg und führt zu einer Serumkonzentration von 150–300 mg/dL. Alpha-1-Antitrypsin ist ein 52-kDa-Molekül, das hauptsächlich in den Hepatozyten produziert und in den Blutkreislauf ausgeschüttet wird. Normalerweise beträgt die tägliche Syntheserate circa 34 mg/kg und führt zu einer Serumkonzentration von 150–300 mg/dL. Alpha-1-Antitrypsin (Synonym: Alpha-1-Proteinasen-Inhibitor) ist in allen Körpergeweben nachweisbar, scheint aber in erster Linie in der Lunge physiologisch bedeutsam zu sein. Aufgrund der großen Kontaktfläche der Lunge mit der Atmungsluft findet in diesem Organ ständig eine Vielzahl von zellulären Abwehrvorgängen statt. Vor allem aus neutrophilen Granulozyten werden dabei hochaktive Proteasen in das umgebende Lungengewebe freigesetzt. 13
 abnormale Sequenzen aufweisen, also ein homozygoter Mangel vorliegt. Normalerweise beträgt die tägliche Syntheserate circa 34 mg/kg und führt zu einer Serumkonzentration von 150–300 mg/dL. Alpha-1-Antitrypsin ist ein 52-kDa-Molekül, das hauptsächlich in den Hepatozyten produziert und in den Blutkreislauf ausgeschüttet wird. Normalerweise beträgt die tägliche Syntheserate circa 34 mg/kg und führt zu einer Serumkonzentration von 150–300 mg/dL. Alpha-1-Antitrypsin (Synonym: Alpha-1-Proteinasen-Inhibitor) ist in allen Körpergeweben nachweisbar, scheint aber in erster Linie in der Lunge physiologisch bedeutsam zu sein. Aufgrund der großen Kontaktfläche der Lunge mit der Atmungsluft findet in diesem Organ ständig eine Vielzahl von zellulären Abwehrvorgängen statt. Vor allem aus neutrophilen Granulozyten werden dabei hochaktive Proteasen in das umgebende Lungengewebe freigesetzt. 13.")
14
Alpha-1-Antitrypsin-Mangel autosomal rezessiv
1 = M – PIMM der gesunde Phänotyp 2 = Z - PIZZ häufigste mit vermindertem AAT einhergehende Genvariante Ca. 100 Genvarianten identifiziert PIZZ - 95% der homozygoten Patienten mit Alpha-1 Antitrypsin-Mangel PI ZZ meist nur 20 bis 50 mg/dL AAT Der Genotyp wird oftmals auch als Erbbild bezeichnet. Alle in den Genen festgelegten Erbinformationen bilden in ihrer Gesamtheit den Genotyp Unter dem Phänotyp versteht man das äußere Erscheinungsbild eines Organismus. Es sind 25 phänotypische Varianten des Alpha-1-PI-Gens durch Mutation am langen Arm des Chromosoms 14 in der Position q bekannt. Eine Charakterisierung ist über Elektrophorese und isoelektrische Fokusierung möglich. Die Bezeichnung erfolgt nach der Wanderungsgeschwindigkeit in der Elektrophorese, wobei folgende Abkürzungenverwandt werden: F=fast, M=medium, S=slow, Z=zero, 0=null. Die mutierten S- und Z-Varianten werden kaum sezerniert. Homozygote Pi00-Träger weisen immer, homozygote PiZZ-Träger fast immer und heterozygote PiSZ- sowie PiZO-Träger oft ein Emphysem auf. Die Pi-ZZ-Variation ist in circa 20 Prozent mit einer Leberzirrhose vergesellschaftet. Auch bei der heterozygoten PiMZ-Variation liegen gehäuft obstruktive Atemwegserkrankungen und eine Leberbeteiligung vor (Berg, Erikson, 1972).
.")
16
Eine der wichtigsten Proteasen ist die neutrophile Elastase, die mit hoher Aktivität das Organ Lunge im Sinne einer „Selbstandauung“ zerstören kann. Beim Gesunden ist das fragile Alveolargewebe durch mehrere Antiproteasen geschützt. Diese Schutzenzyme liegen im Überschuss vor und verursachen eine irreversible Inaktivierung vor allem der neutrophilen Elastase. Alpha-1-Antitrypsin ist quantitativ und auch hinsichtlich der Bindungsaffinität zur neutrophilen Elastase die wichtigste Antiprotease. Aufgrund der großen Kontaktfläche der Lunge mit der Atmungsluft findet in diesem Organ ständig eine Vielzahl von zellulären Abwehrvorgängen statt. Vor allem aus neutrophilen Granulozyten werden dabei hochaktive Proteasen in das umgebende Lungengewebe freigesetzt.

17
Warum ist die Lunge betroffen?
Neutrophile Elastase Antielastase-Schutz Neutrophile Elastase-Last Antielastase-Schutz unzureichend = Alpha-1 Antitrypsin Demzufolge ist die Balance zwischen Proteasen und Antiproteasen in der Lunge von Patienten mit Alpha-1-Antitrypsin-Mangel zur Proteasenseite verschoben. Dieses Ungleichgewicht hat einen langsam progredienten Verlust an Struktur- und Funktionsproteinen zur Folge normal Alpha-1 Antitrypsin-Mangel

18
Warum ist die Lunge betroffen?
Alveolen und Lungengerüst gehören zu den vulnerabelsten Geweben des menschlichen Körpers Durch Proteasen leicht zerstörbar In der Lunge besonders hohe Dichte an Neutrophilen und damit große Belastung durch freisetzbare Proteasen Der antiproteolytische Schutz durch Alpha-1 Antitrypsin ist leicht durch Oxidantien zerstörbar 18

19
Leber Hepatozyten als wesentlicher Syntheseort des Alpha-1-Antitrypsin
AAT vom Typ PIZ wird zwar in normaler Menge synthetisiert Es werden nur 15% aus den Hepatozyten sezerniert. (85% bleiben blockiert) Die Akkumulation führt zur Zellschädigung und letztendlich zur Zirrhose. Die Entstehung von Leberschäden ist weniger gut aufgeklärt als die Pathogenese der Lungenerkrankung. Eine Lebererkrankung wird nur bei einer kleinen Zahl von Patienten mit homozygotem Alpha-1-Antitrypsin-Mangel vom Typ PI*ZZ beobachtet Im Neugeborenenalter entwickelt sich bei einem kleinen Teil der Alpha-1-Antitrypsin-defizienten Patienten ein neonatales hepatisches Syndrom mit Ikterus, der meist nach wenigen Wochen spontan rückläufig ist. Diese Manifestationen sind nicht zwangsläufig mit einer Lebererkrankung in späteren Lebensjahren verbunden. Ab dem Kindes- beziehungsweise Jugendalter können die Symptome einer Hepatitis oder einer Leberzirrhose auftreten. Die abnorm synthetisierten Alpha-1-Antitrypsin-Moleküle der Phänotypen PI*ZZ und einiger sehr seltener M-Varianten (PI*MMalton oder PI*MDuarte) haben die Eigenschaft, durch Polymerisation riesige Molekülverbände zu bilden. Diese Prozesse finden kurze Zeit nach der Synthese noch innerhalb der Leberzelle statt. Histologisch kann eine Akkumulation von Alpha-1-Antitrypsin im rauhen endoplasmatischen Retikulum beobachtet werden. Die gebildeten Molekülkomplexe sind für die Hepatozyten nicht mehr expor- tierbar und führen längerfristig zur Zellschädigung und über eine Fibrose zur Leberzirrhose (10). Bei Alpha-1-Antitrypsinmangel-bedingter Leberzirrhose ist die Inzidenz eines primären Leberzellkarzinoms deutlich erhöht (11). Erwachsenenalter: Prävalenz bei Phänotyp PIZZ 12% Im Alter > 50 Jahre Prävalenz 19% 19
 Die Akkumulation führt zur Zellschädigung und letztendlich zur Zirrhose. Die Entstehung von Leberschäden ist weniger gut aufgeklärt als die Pathogenese der Lungenerkrankung. Eine Lebererkrankung wird nur bei einer kleinen Zahl von Patienten mit homozygotem Alpha-1-Antitrypsin-Mangel vom Typ PI*ZZ beobachtet Im Neugeborenenalter entwickelt sich bei einem kleinen Teil der Alpha-1-Antitrypsin-defizienten Patienten ein neonatales hepatisches Syndrom mit Ikterus, der meist nach wenigen Wochen spontan rückläufig ist. Diese Manifestationen sind nicht zwangsläufig mit einer Lebererkrankung in späteren Lebensjahren verbunden. Ab dem Kindes- beziehungsweise Jugendalter können die Symptome einer Hepatitis oder einer Leberzirrhose auftreten. Die abnorm synthetisierten Alpha-1-Antitrypsin-Moleküle der Phänotypen PI*ZZ und einiger sehr seltener M-Varianten (PI*MMalton oder PI*MDuarte) haben die Eigenschaft, durch Polymerisation riesige Molekülverbände zu bilden. Diese Prozesse finden kurze Zeit nach der Synthese noch innerhalb der Leberzelle statt. Histologisch kann eine Akkumulation von Alpha-1-Antitrypsin im rauhen endoplasmatischen Retikulum beobachtet werden. Die gebildeten Molekülkomplexe sind für die Hepatozyten nicht mehr expor- tierbar und führen längerfristig zur Zellschädigung und über eine Fibrose zur Leberzirrhose (10). Bei Alpha-1-Antitrypsinmangel-bedingter Leberzirrhose ist die Inzidenz eines primären Leberzellkarzinoms deutlich erhöht (11). Erwachsenenalter: Prävalenz bei Phänotyp PIZZ 12% Im Alter > 50 Jahre Prävalenz 19% 19.")
20
Histopathologie Leber
Histopathology PAS (perjod-säure)stained section of liver below show red globular inclusions in hepatocytes. These globules are accumulations of alpha-1-antitrypsin. There maybe slight increase of firbous tissue in portal areas. Rote (PAS-positive) globuläre Einschlusskörper in den Hepatozyten = Akkumulationen von Alpha-1 Antitrypsin
stained section of liver below show red globular inclusions in hepatocytes. These globules are accumulations of alpha-1-antitrypsin. There maybe slight increase of firbous tissue in portal areas. Rote (PAS-positive) globuläre Einschlusskörper in den Hepatozyten = Akkumulationen von Alpha-1 Antitrypsin.")
21
Substitutions-Therapie
Prolastin 60 mg/kg KG/Wo, iv.Gabe FEV1 35 bis 60% PI ZZ, SZ, Z(Null) oder Null/Null Protektive Schwelle – Serumkonzentration von > 0,8 g/l Die Zahl der in Deutschland substituierten Patienten liegt schätzungsweise zwischen 800 und 900. Intravenous augmentation via the infusion of pooled human AAT (alpha-1 antiprotease) Clinical efficacy Limited data are available regarding the clinical efficacy of intravenous AAT. Infusion therapy appears to be safe, well tolerated, and without significant side effects Die Zulassung gilt für die Therapie von Patienten mit den Genotypen PiZZ, PiSZ, PiZ(Null) und Pi(NullNull) und einer mittelgradig eingeschränkten Lungenfunktion mit einem FEV1 im Bereich von 35 bis 60 % des altersentprechenden Sollwerts Beobachtungen an Patienten mit dem Genotyp PiSZ zeigten, dass überwiegend diejenigen mit einer AAT Serum-Konzentration von weniger als 0,8 g/l (11µmol/l) ein Lungenemphysem entwickeln. Diese Konzentration gilt seitdem als „protektive Schwelle“, d.h. als Mindest-Konzentration von AAT ohne wesentlich erhöhtes Risiko für die Entwicklung eines Lungenemphysems [[i]]. Durch intravenöse Applikation von humanem AAT in einer Dosierung von 60 mg/kg Körpergewicht sind AAT Konzentrationen im Serum und in der epithelial lining fluid erreichbar, die für einen Zeitraum von ca. 7 Tagen oberhalb der protektiven Schwellen liegen [i] Crystal RG. Alpha 1-antitrypsin deficiency, emphysema, and liver disease. Genetic basis and strategies for therapy. J Clin Invest. 1990; 85(5): Eine Substitution kann auch bei einem FEV1 Wert von über 65 % des altersentsprechenden Sollwertes begonnen werden, wenn eine ausgeprägte jährliche Reduktion des FEV1 von über 50 ml pro Jahr vorliegt (Evidenzgrad B, vgl. COPD-Leitlinie der Deutschen Gesellschaft für Pneumologie und Beatmungsmedizin e.V. KI Rauchen, selektiver ‚IgA-Mangel 21
 oder Null/Null. Protektive Schwelle – Serumkonzentration von > 0,8 g/l. Die Zahl der in Deutschland substituierten Patienten liegt schätzungsweise zwischen 800 und 900. Intravenous augmentation via the infusion of pooled human AAT (alpha-1 antiprotease) Clinical efficacy Limited data are available regarding the clinical efficacy of intravenous AAT. Infusion therapy appears to be safe, well tolerated, and without significant side effects. Die Zulassung gilt für die Therapie von Patienten mit den Genotypen PiZZ, PiSZ, PiZ(Null) und Pi(NullNull) und einer mittelgradig eingeschränkten Lungenfunktion mit einem FEV1 im Bereich von 35 bis 60 % des altersentprechenden Sollwerts. Beobachtungen an Patienten mit dem Genotyp PiSZ zeigten, dass überwiegend diejenigen mit einer AAT Serum-Konzentration von weniger als 0,8 g/l (11µmol/l) ein Lungenemphysem entwickeln. Diese Konzentration gilt seitdem als „protektive Schwelle , d.h. als Mindest-Konzentration von AAT ohne wesentlich erhöhtes Risiko für die Entwicklung eines Lungenemphysems [[i]]. Durch intravenöse Applikation von humanem AAT in einer Dosierung von 60 mg/kg Körpergewicht sind AAT Konzentrationen im Serum und in der epithelial lining fluid erreichbar, die für einen Zeitraum von ca. 7 Tagen oberhalb der protektiven Schwellen liegen [i] Crystal RG. Alpha 1-antitrypsin deficiency, emphysema, and liver disease. Genetic basis and strategies for therapy. J Clin Invest. 1990; 85(5): Eine Substitution kann auch bei einem FEV1 Wert von über 65 % des altersentsprechenden Sollwertes begonnen werden, wenn eine ausgeprägte jährliche Reduktion des FEV1 von über 50 ml pro Jahr vorliegt (Evidenzgrad B, vgl. COPD-Leitlinie der Deutschen Gesellschaft für Pneumologie und Beatmungsmedizin e.V. KI Rauchen, selektiver ‚IgA-Mangel. 21.")
22
Kaplan–Meier cumulative mortality curves based on all eligible patients and deaths, plotted for subjects with initial FEV1 < 50% predicted and for those with initial FEV1 ⩾ 50% predicted. In each plot, separate curves are shown for subjects classified as never receiving (thick solid line), partly receiving (dotted line), and always receiving (narrow solid line) augmentation therapy. The log-rank p value presented is for a comparison of the subjects never receiving therapy with the combined group of subjects partly or always receiving therapy. (A and B) Kaplan–Meier plots of survival from time of enrollment using data from all subjects. (C and D) Similar analysis, but restricted to those subjects who had follow-up contact for at least 6 mo after enrolling in the Registry. Survival and FEV1 Decline in Individuals with Severe Deficiency of α1 Antitrypsin Am. J. Respir. Crit. Care Med.July1,1998 vol.158 no.1,49-59 22

23
Wencker M, Fuhrmann B, Banik N, et al
Wencker M, Fuhrmann B, Banik N, et al. Longitudinal follow-up of patients with alpha(1)-protease inhibitor deficiency before and during therapy with IV alpha(1)-protease inhibitor. Chest 2001;119:737–44. Wolfgang Gleiber 23
-protease inhibitor deficiency before and during therapy with IV alpha(1)-protease inhibitor. Chest 2001;119:737–44. Wolfgang Gleiber. 23.")
24
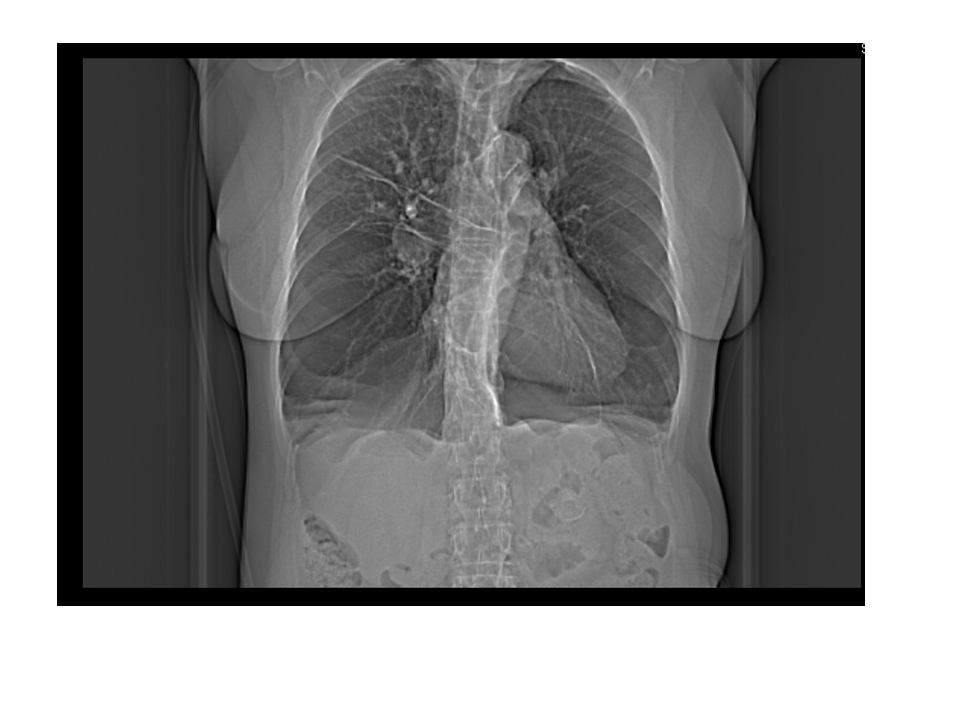
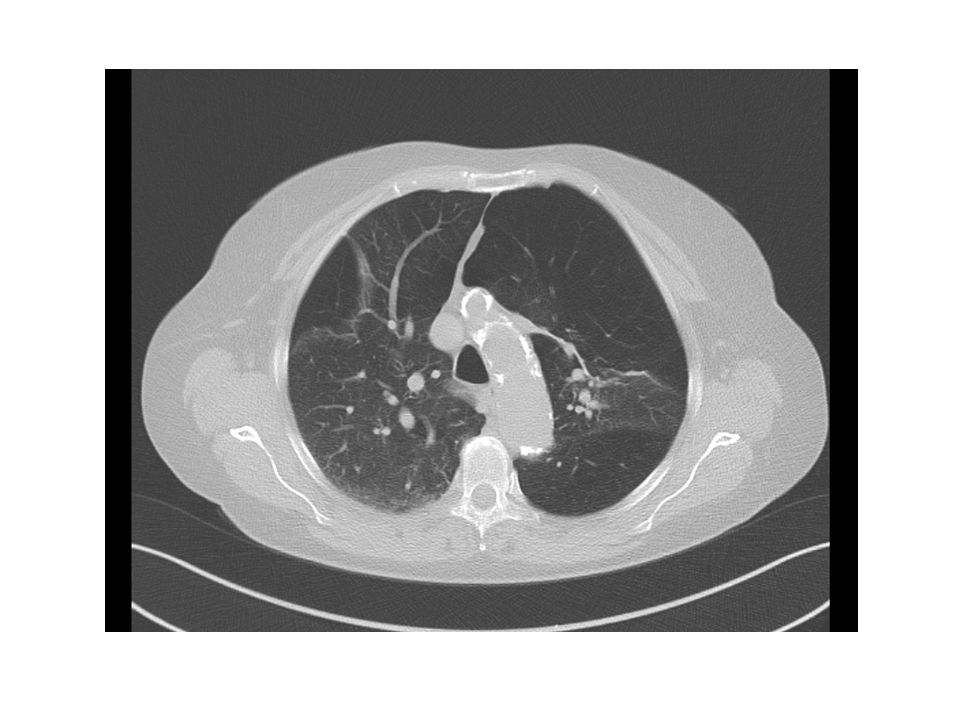
Lungenemphysem Panlobulär – Diffuser Befall, im Lobulus werden alle Alveolen zerstört. Vorzugsweise sind die basalen Lungenabschnitte betroffen. Typisch für den Alpha-1 Antitrypsinmangel. Zentrilobulär – von den Bronchioli respiratorii ausgehend. Häufigste Form, Zigarettenraucher, vorzugsweise in den apikalen Abschnitten

26
Prolastin: 60 mg/kg KG/Wo
More recently, the EXAcerbations and CT scan as Lung Endpoints (EXACTLE) study (77 patients studied over months), using a similar placebo-controlled trial design of IV AAT, explored CT densitometry as the primary outcome Die EXACTLE-Studie dokumentierte bei 77 Patienten mit schwerem Alpha-1-Antitrypsin-Mangel über 2 bis 2,5 Jahre den Verlauf des Lungenemphysems mithilfe der CT-Densitometrie Mit dieser Methode konnte man einen zunehmenden Verlust von Lungengewebe erfassen Mit der Hounsfield-Skala wird in der Computertomographie (CT) die Abschwächung von Röntgenstrahlung in Gewebe beschrieben und für die Darstellung in einem Graustufenbild benutzt Luft absorbiert Röntgenstrahlung nahezu gar nicht und hat definitionsgemäß eine CT-Zahl von −1000 HU. Wasser hat gemäß der Definition 0 HU Knochen haben, je nach Dichte, Werte von 500 bis 1500 HU Exploring the role of CT densitometry: a randomised study of augmentation therapy in a1-antitrypsin deficiency (EXACTLE) Dirksen et al., Eur Resp J 2009 26
 study (77 patients studied over months), using a similar placebo-controlled trial design of IV AAT, explored CT densitometry as the primary outcome Die EXACTLE-Studie dokumentierte bei 77 Patienten mit schwerem Alpha-1-Antitrypsin-Mangel über 2 bis 2,5 Jahre den Verlauf des Lungenemphysems mithilfe der CT-Densitometrie Mit dieser Methode konnte man einen zunehmenden Verlust von Lungengewebe erfassen. Mit der Hounsfield-Skala wird in der Computertomographie (CT) die Abschwächung von Röntgenstrahlung in Gewebe beschrieben und für die Darstellung in einem Graustufenbild benutzt. Luft absorbiert Röntgenstrahlung nahezu gar nicht und hat definitionsgemäß eine CT-Zahl von −1000 HU. Wasser hat gemäß der Definition 0 HU Knochen haben, je nach Dichte, Werte von 500 bis 1500 HU. Exploring the role of CT densitometry: a randomised study of augmentation therapy in a1-antitrypsin deficiency (EXACTLE) Dirksen et al., Eur Resp J")
27
Data analysis and CT densitometry
The rate of emphysema progression was determined by change in lung density measured by whole lung CT scan, and reported as the annual change in the 15th percentile lung density (PD15) (determined from the endpoint in the original trials). The PD15 value is extracted from the frequency histogram of lung voxels and is the density value (g•L-1) at which 15% of the voxels have lower densities [9,10] (Figure 2). This analysis combines the raw data from both trials, thereby increasing the numbers of patients and the robustness of the analysis. although the exacerbation frequency and pulmonary function were not affected 27
 (determined from the endpoint in the original trials). The PD15 value is extracted from the frequency histogram of lung voxels and is the density value (g•L-1) at which 15% of the voxels have lower densities [9,10] (Figure 2). This analysis combines the raw data from both trials, thereby increasing the numbers of patients and the robustness of the analysis. although the exacerbation frequency and pulmonary function were not affected. 27.")
28
„Alpha-1-kit“ Am Deutschen Alpha-1-Antitrypsin-Zentrum an der Philipps-Universität Marburg besteht die Möglichkeit, durch Einsendung eines so genannten „Alpha-1-kit“ den Phänotyp beziehungsweise den Genotyp im Rahmen eines Pilotprojekts kostenlos bestimmen zu lassen. Alpha-1-kits sind spezielle Filterpapierstreifen, auf die wenige Bluttropfen eines Probanden aufgebracht werden. Ähnlich wie beim Neugeborenen-Screening kann daraus die genannte Diagnostik erfolgen. Daraus folgt die Empfehlung, zeitgleich zur Alpha-1-Antitrypsin-Bestimmung das C-reaktive Protein (CrP) zu messen, um die genannte Situation mit pseudonormalem Alpha-1-Antitrypsin-Spiegel ausschließen zu können. 28
 zu messen, um die genannte Situation mit pseudonormalem Alpha-1-Antitrypsin-Spiegel ausschließen zu können. 28.")
29
International Study Evaluating the Safety and Efficacy of Inhaled, Human, Alpha-1 Antitrypsin (AAT) in Alpha-1 Antitrypsin Deficient Patients With Emphysema Kamada-Protokoll Inclusion Patient with record of congenital AAT deficiency of phenotype PiZZ (homozygote) or other rare phenotypes related to AAT deficiency and with AAT serum level ≤ 11 micromole. Gegen Placebo 29
 or other rare phenotypes related to AAT deficiency and with AAT serum level ≤ 11 micromole. Gegen Placebo. 29.")
30
Endoskopische Lungenvolumenreduktion bei alpha1-Antitrypsinmangel
Schwergradiges Lungenemphysem FEV1 < 45 % , RV > 150 % , TLC > 100 % maximal medikamentöse Therapie gemäß GOLD AAT-Mangel AAT-Spiegel < 80 mg/dl, Genotyp: PiSZ, PiZZ, Pi(0/0) Blutgase pO2 > 60 mmHg unter 4 l O2 pCO2 < 50 mmHg nativ in Ruhe Heterogenität Heterogenes Lungenemphysem, welches bereits vor Studieneinschluss durch eine HCRT des Thorax und eine Perfusionsszintigraphie nachgewiesen wurde Bestätigung der Heterogenität durch computergestützte Emphysemquantifizierung (YACTA®) Patientenalter > 30 Jahre Stabile COPD Nicht- bzw. Exraucher 30
 Blutgase pO2 > 60 mmHg unter 4 l O2. pCO2 < 50 mmHg nativ in Ruhe. Heterogenität Heterogenes Lungenemphysem, welches bereits vor Studieneinschluss durch eine HCRT des Thorax und eine Perfusionsszintigraphie nachgewiesen wurde. Bestätigung der Heterogenität durch computergestützte Emphysemquantifizierung (YACTA®) Patientenalter > 30 Jahre. Stabile COPD. Nicht- bzw. Exraucher. 30.")
31
Vielen Dank für Ihre Aufmerksamkeit !
Wolfgang Gleiber Medizinische Klinik 1 Schwerpunkt Pneumologie/Allergologie Johann Wolfgang Goethe-Universität Frankfurt am Main 31

Ähnliche Präsentationen
















 und Nichtinvasive Beatmung (NIV)>")






