Präsentation herunterladen
Die Präsentation wird geladen. Bitte warten

1
Apoptosemechanismen und neurodegenerative Erkrankungen
Die Alzheimer Demenz Dr. Claudia Götz

2
Übersicht – Die Alzheimer Demenz
Die Entdeckung der Krankheit Charakteristika der Alzheimer – Krankheit „Alzheimer – Hirn“ und gesundes Hirn im Vergleich Amyloidplaques im „Alzheimer – Hirn“ Ursachen, Risikofaktoren Behandlungsstrategien Molekulare Veränderungen in der Alzheimer-Demenz Ab tau Resultate der molekularen Veränderungen in der Alzheimer-Demenz

3
Übersicht - Apoptose Formen des Zelltods - Apoptose
Nachweismöglichkeiten der Apoptose auf molekularer Ebene Notwendigkeit der Apoptose Phasen der Apoptose Caspasen und Apoptose Zwei Wege der Apoptoseinduktion Der mitochondriale Weg der Apoptose Der Todesrezeptor-Weg der Apoptose Übersicht über die bekannten Apoptosemechanismen Deregulierte Apoptose

4
Übersicht – Alzheimer und Apoptose
Apoptose und AD Bedeutung des Ab für die Apoptose in AD Bedeutung des tau – Proteins für die Apoptose in AD Caspase-Spaltung von tau führt zur Ausbildung von NFT Zusammenfassung der molekularen Vorgänge bei der Apoptose in AD

5
Die Entdeckung der Krankheit
Erstbeschreibung der Krankheit durch den Würzburger Arzt Alois Alzheimer im Jahr Er schrieb über seine Patientin Auguste D.: „Eine Frau von 51 Jahren zeigte als erste auffällige Krankheitserscheinung Eifersuchtsideen gegen den Mann. Bald machte sich eine rasch zunehmende Gedächtnisschwäche bemerkbar, sie fand sich in ihrer Wohnung nicht mehr zurecht, schleppte Gegenstände hin und her, versteckte sie, zuweilen glaubte sie, man wolle sie umbringen und begann, laut zu schreien.“ Auguste D. starb fünf Jahre später. Als Alzheimer ihr Gehirn untersuchte, fand er darin steinharte Ablagerungen, die sogenannten Plaques. Schon Rudolf Virchow hatte diese Plaques in Gehirnen Verstorbener entdeckt, nannte sie Amyloid-Plaques, konnte sie aber nicht in Zusammenhang bringen mit dem Verlust der geistigen Leistungsfähigkeit. Unter der Alzheimer Krankheit oder Morbus Alzheimer oder Demenz vom Alzheimer – Typ versteht man den Verfall der geistigen Leistungsfähigkeit. Die Alzheimer-Demenz zeichnet sich vor allem aus durch eine anfängliche Gedächtnisschwäche, die im Verlauf zunimmt und zum Totalverlust der Urteilsfähigkeit und der Persönlichkeit führen kann. Direkt nach Schlaganfällen ist die Alzheimer – Demenz die häufigste schwere Störung der Hirnfunktion im Alter. 20 – 30 Mio Menschen WW Alois Alzheimer und seine Patientin Auguste D.

6
Charakteristika der Alzheimer - Krankheit
Störungen des Kurzzeitgedächtnisses Denkschwierigkeiten Sprachstörungen Depressionen Eingeschränktes Urteilsvermögen Wahnvorstellungen Persönlichkeitsveränderungen Verschlechterung des Kurzzeitgedächtnisses als erstes Symptom ist schon im Alter von 60 – 70 Jahren zu beobachten. Die Erinnerungen an die Jugend (biographisches Gedächtnis) sind oft noch sehr präsent. Im Verlauf der Progression der Krankheit kommen die übrigen Symptome hinzu. Verhaltensänderungen wie Verwirrtheit, Angst, Unruhe, Aggressivität. Den Betroffenen fällt es schwer, Dinge und Personen wiederzuerkennen; sie leben in der Vergangenheit. Alltagsfähigkeiten wie Ankleiden, Essenszubereitung oder Einkaufen können sie nicht mehr bewältigen. Schließlich verlieren sie die Kontrolle über ihre Körperfunktionen. Im Endstadium verstummen die Betroffenen oft, sind bettlägerig und völlig auf die Hilfe Fremder angewiesen.
 sind oft noch sehr präsent. Im Verlauf der Progression der Krankheit kommen die übrigen Symptome hinzu. Verhaltensänderungen wie Verwirrtheit, Angst, Unruhe, Aggressivität. Den Betroffenen fällt es schwer, Dinge und Personen wiederzuerkennen; sie leben in der Vergangenheit. Alltagsfähigkeiten wie Ankleiden, Essenszubereitung oder Einkaufen können sie nicht mehr bewältigen. Schließlich verlieren sie die Kontrolle über ihre Körperfunktionen. Im Endstadium verstummen die Betroffenen oft, sind bettlägerig und völlig auf die Hilfe Fremder angewiesen.")
7
„Alzheimer – Hirn“ und gesundes Hirn im Vergleich
Links: Alzheimer Hirn, rechts gesundes Hirn Sichtbar: Schrumpfung des Gehirns, da Nervenzellen absterben, Nervenverschaltungen (Synapsen) gehen verloren, v.a. in Regionen, die an der Entwicklung von Sprache, Gedächtnis und Denkfähigkeit beteiligt sind. Insgesamt wird der Stoffwechsel reduziert
 gehen verloren, v.a. in Regionen, die an der Entwicklung von Sprache, Gedächtnis und Denkfähigkeit beteiligt sind. Insgesamt wird der Stoffwechsel reduziert.")
8
„Alzheimer – Hirn“ und gesundes Hirn im Vergleich
Links: Alzheimer Hirn, rechts gesundes Hirn Sichtbar: Schrumpfung des Gehirns, da Nervenzellen absterben, Nervenverschaltungen (Synapsen) gehen verloren, v.a. in Regionen, die an der Entwicklung von Sprache, Gedächtnis und Denkfähigkeit beteiligt sind. Insgesamt wird der Stoffwechsel reduziert
 gehen verloren, v.a. in Regionen, die an der Entwicklung von Sprache, Gedächtnis und Denkfähigkeit beteiligt sind. Insgesamt wird der Stoffwechsel reduziert.")
9
Amyloidplaques im „Alzheimer – Hirn“
Alzheimer – Krankheit ist schwer zu erkennen (CT, EEG, Kernspin-Tomographie, neuropsychologische Tests..) Sicherheit in der Diagnose erhält man erst nach dem Tod anhand der Amyloidplaques (Silberfärbung) = Ablagerungen aus Proteinfragmenten in Form extrazellulärer, kugelförmiger Ablagerungen (Plaques) (im Bild zu sehen) oder intrazellulär in Form von Fibrillen (neurofibrillary tangles NFT) Amyloid: Stärke-ähnlich, Namensgeber: R. Virchow Anfärbbarkeit mit Perjodsäure
 Sicherheit in der Diagnose erhält man erst nach dem Tod anhand der Amyloidplaques (Silberfärbung) = Ablagerungen aus Proteinfragmenten in Form extrazellulärer, kugelförmiger Ablagerungen (Plaques) (im Bild zu sehen) oder intrazellulär in Form von Fibrillen (neurofibrillary tangles NFT) Amyloid: Stärke-ähnlich, Namensgeber: R. Virchow Anfärbbarkeit mit Perjodsäure.")
10
Ursachen, Risikofaktoren
Ursache unbekannt Ausnahme: in seltenen Fällen (10%) Genveränderungen im APP-Protein und Präsenilinen Risikofaktoren: Apo E – Variante (Cholesterintransport) Lebensalter Oxidativer Stress Nikotin, Alkohol Diabetes, Schilddrüsenunterfunktion, Bluthochdruck Genetisch bedingte Alzheimer Erkrankung beginnt schon vor dem 60. Lebensjahr (durchaus auch schon im dritten Lebensjahrzehnt) Durch Mutationen im APP können sich vermehrt schädliche Spaltprodukte bilden, der Hauptbestandteil der sog. Plaques Etwa 20 missense Mut bekannt frühe u aggressive Form Trisomie 21 führt schon ab dem 40. Lebensjahr zur Demenz ( Gen für APP liegt ebenfalls auf Chromosom 21 Überexpression) Mutierte Präseniline (wahrsch. Bestandteil der gamma-Sekretase) können die Aktivität von Molekülen erhöhen, die das APP zerschneiden. Etwa 150 missense Mut bekannt aggressive Frühform Mutationen sprechen für Amyloidhypothese, da alle Mutationen die zu autosomal dom. AD führen entweder im APP Gen oder der Ab produzierenden Protease liegen. ApoE wichtigster Risikofaktor für späte Form. drei Varianten vor: e3: 70-80% der Bev., e4: 10-15%, e2: 5-15%; unter AD-Patienten: 45% Gehirn 2. Ort neben Leber wo ApoE produziert wird, v.a.Astrocyten protektive Rolle, Expression steigt stark (250x) nach Verletzung an Homozygot ApoE4 erhöht das Krankheitsrisiko im Durchschnitt um das 9 – 10x, eine andere Variante (ApoE2) senkt das Risiko. ApoE4 fördert die Ab – Aggregation und ist mit Ab in den senilen Plaques vergesellschaftet. ApoE4 instabiler u. proteolyseanfälliger als E3 Modell: Ab neurotoxiscj ApoE expression, für E4: Instabilität Neurodegeneration E3 scheint Ab-Abbau stärker zu fördern als E4 E3 kann im Ggs. Zu E4 als Antioxidans wirken (Cys) Bildung von Sauerstoffradikalen – beim älteren Menschen sowieso erhöht - ist beim Alzheimer – Patienten noch zusätzlich stärker. (u.a. durch Anreicherung von Metallionen zwischen den Zellen; Ab kann redoxaktive Metallionen mit einer hohen Aktivität binden)
 Genveränderungen im APP-Protein und Präsenilinen. Risikofaktoren: Apo E – Variante (Cholesterintransport) Lebensalter. Oxidativer Stress. Nikotin, Alkohol. Diabetes, Schilddrüsenunterfunktion, Bluthochdruck. Genetisch bedingte Alzheimer Erkrankung beginnt schon vor dem 60. Lebensjahr (durchaus auch schon im dritten Lebensjahrzehnt) Durch Mutationen im APP können sich vermehrt schädliche Spaltprodukte bilden, der Hauptbestandteil der sog. Plaques. Etwa 20 missense Mut bekannt frühe u aggressive Form. Trisomie 21 führt schon ab dem 40. Lebensjahr zur Demenz ( Gen für APP liegt ebenfalls auf Chromosom 21 Überexpression) Mutierte Präseniline (wahrsch. Bestandteil der gamma-Sekretase) können die Aktivität von Molekülen erhöhen, die das APP zerschneiden. Etwa 150 missense Mut bekannt aggressive Frühform. Mutationen sprechen für Amyloidhypothese, da alle Mutationen die zu autosomal dom. AD führen entweder im APP Gen oder der Ab produzierenden Protease liegen. ApoE wichtigster Risikofaktor für späte Form. drei Varianten vor: e3: 70-80% der Bev., e4: 10-15%, e2: 5-15%; unter AD-Patienten: 45% Gehirn 2. Ort neben Leber wo ApoE produziert wird, v.a.Astrocyten protektive Rolle, Expression steigt stark (250x) nach Verletzung an. Homozygot ApoE4 erhöht das Krankheitsrisiko im Durchschnitt um das 9 – 10x, eine andere Variante (ApoE2) senkt das Risiko. ApoE4 fördert die Ab – Aggregation und ist mit Ab in den senilen Plaques vergesellschaftet. ApoE4 instabiler u. proteolyseanfälliger als E3. Modell: Ab neurotoxiscj ApoE expression, für E4: Instabilität Neurodegeneration. E3 scheint Ab-Abbau stärker zu fördern als E4. E3 kann im Ggs. Zu E4 als Antioxidans wirken (Cys) Bildung von Sauerstoffradikalen – beim älteren Menschen sowieso erhöht - ist beim Alzheimer – Patienten noch zusätzlich stärker. (u.a. durch Anreicherung von Metallionen zwischen den Zellen; Ab kann redoxaktive Metallionen mit einer hohen Aktivität binden)")
11
Behandlungsstrategien
Keine Heilung möglich, nur Retardierung Acetylcholinesterase – Hemmer NMDA – Antagonisten Antioxidantien (Vitamin E) Kupfer Ergotherapie, Gedächtnistraining Acetylcholinesterase-Hemmer: Insgesamt weniger Neuotransmitter gebildet. Versuch den Austausch der Signale zwischen den überlebenden Zellen zu verbessern; Steigerung der Verfügbarkeit des Acetylcholin. Nur im Frühstadium der Erkrankung sinnvoll lediglich Verbesserung der kognitiven Fähigkeiten, Krankheitsfortschritt wird nicht aufgehalten! Andere Transmitter sind ungleichmäßig häufig im Gehirn verteilt und können zu einer Überreizung der Nervenzellen führen. NMDA-Rezeptoren gehören zu den ionotropen Glutamat-Rezeptoren und kommen vor allem im Zentralnervensystem vor. Sie sind nach dem ebenfalls wirksamen selektiven Agonisten N-Methyl-D-Aspartat benannt. Mit NMDA-Antagonisten wird die Nervenüberreizung durch Glutamat verhindert. Kupfer verhindert die Bildung von hochmolekularen Aggregaten des Ab; das Proteinfragment bleibt monomer (auch Studie hier in Homburg mit vielversprechenden Ergebnissen)
 Kupfer. Ergotherapie, Gedächtnistraining. Acetylcholinesterase-Hemmer: Insgesamt weniger Neuotransmitter gebildet. Versuch den Austausch der Signale zwischen den überlebenden Zellen zu verbessern; Steigerung der Verfügbarkeit des Acetylcholin. Nur im Frühstadium der Erkrankung sinnvoll lediglich Verbesserung der kognitiven Fähigkeiten, Krankheitsfortschritt wird nicht aufgehalten! Andere Transmitter sind ungleichmäßig häufig im Gehirn verteilt und können zu einer Überreizung der Nervenzellen führen. NMDA-Rezeptoren gehören zu den ionotropen Glutamat-Rezeptoren und kommen vor allem im Zentralnervensystem vor. Sie sind nach dem ebenfalls wirksamen selektiven Agonisten N-Methyl-D-Aspartat benannt. Mit NMDA-Antagonisten wird die Nervenüberreizung durch Glutamat verhindert. Kupfer verhindert die Bildung von hochmolekularen Aggregaten des Ab; das Proteinfragment bleibt monomer (auch Studie hier in Homburg mit vielversprechenden Ergebnissen)")
12
Molekulare Veränderungen in der Alzheimer-Demenz - Ab
APP: Rezeptorprotein mit Transmembrandomäne, 770 AS Wahrsch. Fkt als autokriner Faktor Neuritenwachstum, neuronalen Adhesion, Bildung des Axons beteiligt Rezeptor für Kinesin? Transkriptionsfaktor? genaue Funktion unbekannt. a-Sekretase wird APP in eine lösliche Form überführt und kann im Plasma nachgewiesen werden. b- und g-Sekretase erzeugen ebenfalls ein lösliches APP und das Ab-Protein, das zur Bildung der Plaques führt. (auch beim nicht-dementen Patienten in geringen Konzentrationen nachweisbar) Reifung auf sekretorischem Weg zur Plasmamembran (a-sekretase) und erneute Aufnahme durch Endocytose b- und g-Sekretasen wahrsch. im Endosom intrazelluläre Ablagerung mögl. Ab40 normales zelluläres Produkt, Fkt unbekannt (mehr als 95% des Ab) Ab42 „langes“ Ab (weniger als 5%) wahrsch. für die Initiierung der Oligomerbildung verantwortlich Amyloidkaskade-Hypothese Mutationen im APP, die zu einer frühen Form der Demenz führen sind hauptsächlich in diesen Schnittstellen zu finden. Extrazelluläre (Plaques) und intrazelluläre Ablagerung des Ab
 Reifung auf sekretorischem Weg zur Plasmamembran (a-sekretase) und erneute Aufnahme durch Endocytose b- und g-Sekretasen wahrsch. im Endosom intrazelluläre Ablagerung mögl. Ab40 normales zelluläres Produkt, Fkt unbekannt (mehr als 95% des Ab) Ab42 „langes Ab (weniger als 5%) wahrsch. für die Initiierung der Oligomerbildung verantwortlich Amyloidkaskade-Hypothese. Mutationen im APP, die zu einer frühen Form der Demenz führen sind hauptsächlich in diesen Schnittstellen zu finden. Extrazelluläre (Plaques) und intrazelluläre Ablagerung des Ab.")
13
Molekulare Veränderungen in der Alzheimer- Demenz
Amyloide besitzen typ. Exponentielles Wachstumsverhalten = „Seeding“ einige weinige Fibrillen die sich gebildet haben instuieren die Fehlfaltung weiterer amyloider Pepptide Plaquebildung Ab-Oligomere wirken neurotoxisch, Plaques relativ inert APP intracellular domain Extrazelluläre (Plaques) und intrazelluläre Ablagerung des Ab
 und intrazelluläre Ablagerung des Ab.")
14
Molekulare Veränderungen in der Alzheimer-Demenz - Tau
PHF Intrazelluläre Ablagerung Mikrotubuli-assoziiertes Protein, füördert Zusammenbau und Stabilität der Mikrotubuli, Beteiligunng an der Ausprägung und der Aufrechterhaltung der Polarität beteiligt. Der C-Terminus bindet axonale Mikrotubuli, der N-Terminus Plasmamembrankomponenten der Nervenzellen. Ist an Transportvorgängen entlang der Mikrotubuli beteiligt. Tau-Veränderungen (Mutationen) Tauopathien Neuronale Degeneration, Demenz, Tod Ausbildung von NFTs aber OHNE Ab-Plaques sind aber Def.-Kriterium für AD Tau kann nicht ursächlich für AD Entstehung sein Mögl. Weg auf dem Ab zur Bildung von NFT durch Tau-Phosphorylierung führt Gegenstand derzeitiger Untersuchung Mögl. Rolle für ApoE: gesteigerte Tau Phosphorylierung in transgenen Mäusen mit ApoE4 E3 bindet an Tau verhindert Hyperphosphorylierung und MT-Destabilisierung
 Tauopathien. Neuronale Degeneration, Demenz, Tod. Ausbildung von NFTs aber OHNE Ab-Plaques sind aber Def.-Kriterium für AD. Tau kann nicht ursächlich für AD Entstehung sein. Mögl. Weg auf dem Ab zur Bildung von NFT durch Tau-Phosphorylierung führt Gegenstand derzeitiger Untersuchung. Mögl. Rolle für ApoE: gesteigerte Tau Phosphorylierung in transgenen Mäusen mit ApoE4. E3 bindet an Tau verhindert Hyperphosphorylierung und MT-Destabilisierung.")
15
Resultate der molekularen Veränderungen in der Alzheimer-Demenz
erschwerte Kommunikation zwischen den Nervenzellen vermehrtes Absterben der Nervenzellen durch Apoptose

16
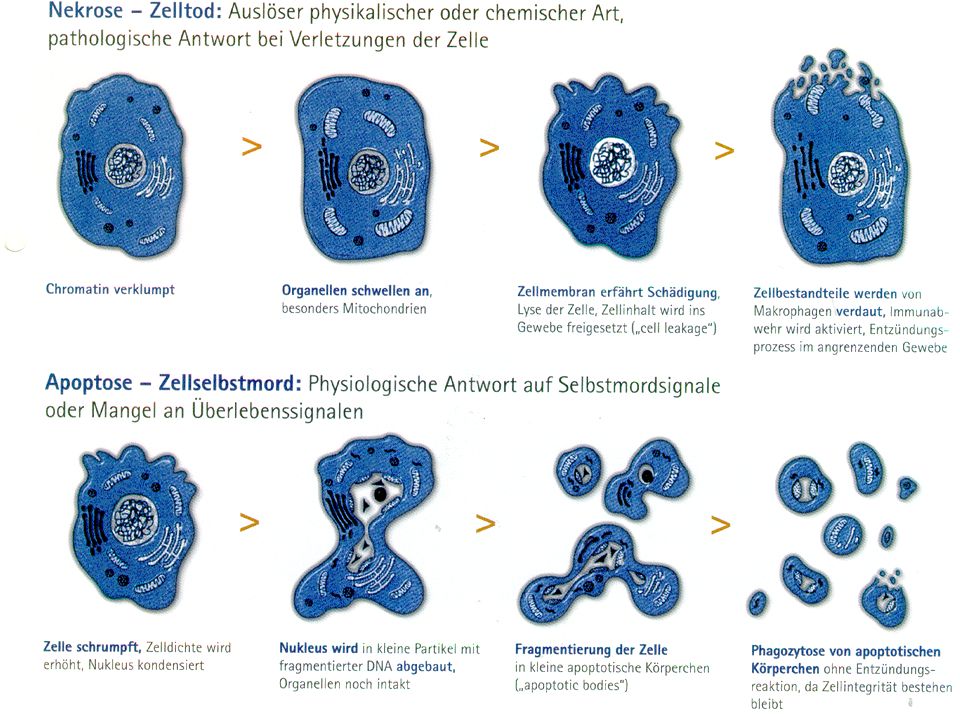
Formen des Zelltods

18
Nachweismöglichkeiten der Apoptose auf molekularer Ebene
Nachweis der DNA-Fragmentierung

19
Nachweismöglichkeiten der Apoptose auf molekularer Ebene
Poly-ADP-Ribosepolymerase PARP Aktive Caspase 3 116 kDa 89 kDa 27 kDa Vollängen-PARP PARP-Spaltprodukt µM Emodin Indirekter Nachweis der Caspase 3 Aktivität durch Spaltung der PARP

20
Notwendigkeit der Apoptose

21
Phasen der Apoptose

22
Caspasen und Apoptose Cystein-Aspartat-Proteasen
Initiatorcaspasen: Caspase 8 Caspase 10 Caspase 9 Caspase 1 Caspase 5 Caspase 4 Caspase 12 Caspase 2 Spaltung und Aktivierung der Pro-Caspasen durch limitierte Proteolyse Executionscaspasen Caspase 3 Caspase 7 Caspase 6 Spaltung und Inaktivierung zellulärer Proteine (z.B. Parp, Actin, aber auch APP. Präseniline, tau...) aber auch Aktivierung von DNasen ( Fragmentierung von DNA)
 aber auch Aktivierung von DNasen ( Fragmentierung von DNA)")
23
Zwei Wege der Apoptoseinduktion

24
Der mitochondriale Weg der Apoptose
Programmed Cell Death Programmed cell death (PCD), or apoptosis, can be triggered by a wide range of stimuli, including cell surface receptors like Fas or tumor necrosis factor receptor 1 (TNFR1). It constitutes a system for the removal of unnecessary, aged, or damaged cells that are regulated by the interplay of proapoptotic and antiapoptotic proteins of the Bcl-2 family. The proapoptotic proteins Bax, Bad, Bid, Bik, and Bim contain an a-helical BH3 death domain that fits the hydrophobic BH3 binding pocket on the antiapoptotic proteins Bcl-2 and Bcl-XL, forming heterodimers that block the survival-promoting activity of Bcl-2 and Bcl-XL. Thus, the relative abundance of proapoptotic and antiapoptotic proteins determines the susceptibility of the cell to programmed death. The proapoptotic proteins act at the surface of the mitochondrial membrane to decrease the mitochondrial transmembrane potential and promote leakage of cytochrome c. In the presence of dATP, cytochrome c complexes with and activates Apaf-1. Activated Apaf-1 binds to downstream caspases, such as pro-caspase-9, and processes them into proteolytically active forms. This begins a caspase cascade resulting in apoptosis. Smac/Diablo is released from the mitochondria and blocks IAP proteins which normally interact with caspase-9 to inhibit apoptosis. The lower panel shows the conserved apoptotic pathway in C. elegans. CED-3 encodes a caspase whose function is facilitated by CED-4, which is highly similar to Apaf-1. CED-4 function is blocked by CED-9, which protects cells against apoptosis and is similar to the human antiapoptotic protein Bcl-2. CED-9 activity is inhibited by EGL-1, which is similar to the proapoptotic Bcl-2 family members.
, or apoptosis, can be triggered by a wide range of stimuli, including cell surface receptors like Fas or tumor necrosis factor receptor 1 (TNFR1). It constitutes a system for the removal of unnecessary, aged, or damaged cells that are regulated by the interplay of proapoptotic and antiapoptotic proteins of the Bcl-2 family. The proapoptotic proteins Bax, Bad, Bid, Bik, and Bim contain an a-helical BH3 death domain that fits the hydrophobic BH3 binding pocket on the antiapoptotic proteins Bcl-2 and Bcl-XL, forming heterodimers that block the survival-promoting activity of Bcl-2 and Bcl-XL. Thus, the relative abundance of proapoptotic and antiapoptotic proteins determines the susceptibility of the cell to programmed death. The proapoptotic proteins act at the surface of the mitochondrial membrane to decrease the mitochondrial transmembrane potential and promote leakage of cytochrome c. In the presence of dATP, cytochrome c complexes with and activates Apaf-1. Activated Apaf-1 binds to downstream caspases, such as pro-caspase-9, and processes them into proteolytically active forms. This begins a caspase cascade resulting in apoptosis. Smac/Diablo is released from the mitochondria and blocks IAP proteins which normally interact with caspase-9 to inhibit apoptosis. The lower panel shows the conserved apoptotic pathway in C. elegans. CED-3 encodes a caspase whose function is facilitated by CED-4, which is highly similar to Apaf-1. CED-4 function is blocked by CED-9, which protects cells against apoptosis and is similar to the human antiapoptotic protein Bcl-2. CED-9 activity is inhibited by EGL-1, which is similar to the proapoptotic Bcl-2 family members.")
25
Der Todesrezeptor-Weg der Apoptose
Fas Signaling Pathway Fas/APO-1/CD95 (36 kDa) is a member of the tumor necrosis factor (TNF) receptor superfamily, a family of transmembrane receptors that also includes p75 neurotrophin receptor, TNFRI, and a variety of other cell surface receptors. Fas has been shown to be an important mediator of apoptotic cell death, as well as being involved in inflammation. Binding of the Fas ligand (Fas-L) induces trimerization of Fas in the target cell membrane. Activation of Fas causes the recruitment of Fas-associated protein with death domain (FADD) via interactions between the death domains of Fas and FADD. Pro-caspase-8 binds to Fas-bound FADD via interactions between the death effector domains (DED) of FADD and pro-caspase-8 leading to the activation of caspase-8. Activated caspase-8 cleaves (activates) nine other pro-caspases, in effect beginning a caspase cascade that ultimately leads to apoptosis. Caspases cleave nuclear lamins, causing the nucleus to break down and lose its normal structure. Fas-induced apoptosis can be effectively blocked at several stages by either FLICE-inhibitory protein (FLIP), by Bcl-2, or by the cytokine response modifier A (CrmA). Moreover, caspase-8 can activate Bid which is then able to associate with the mitochondria and promote leakage of cytochrome c. In the presence of dATP, cytochrome c complexes with and activates Apaf-1. Activated Apaf-1 binds to downstream caspases, such as pro-caspase-9, and processes them into proteolytically active forms. This begins a caspase cascade resulting in apoptosis. In addition, Smac/Diablo is released from the mitochondria and blocks IAP proteins which normally interact with caspase-9 to inhibit apoptosis.
 is a member of the tumor necrosis factor (TNF) receptor superfamily, a family of transmembrane receptors that also includes p75 neurotrophin receptor, TNFRI, and a variety of other cell surface receptors. Fas has been shown to be an important mediator of apoptotic cell death, as well as being involved in inflammation. Binding of the Fas ligand (Fas-L) induces trimerization of Fas in the target cell membrane. Activation of Fas causes the recruitment of Fas-associated protein with death domain (FADD) via interactions between the death domains of Fas and FADD. Pro-caspase-8 binds to Fas-bound FADD via interactions between the death effector domains (DED) of FADD and pro-caspase-8 leading to the activation of caspase-8. Activated caspase-8 cleaves (activates) nine other pro-caspases, in effect beginning a caspase cascade that ultimately leads to apoptosis. Caspases cleave nuclear lamins, causing the nucleus to break down and lose its normal structure. Fas-induced apoptosis can be effectively blocked at several stages by either FLICE-inhibitory protein (FLIP), by Bcl-2, or by the cytokine response modifier A (CrmA). Moreover, caspase-8 can activate Bid which is then able to associate with the mitochondria and promote leakage of cytochrome c. In the presence of dATP, cytochrome c complexes with and activates Apaf-1. Activated Apaf-1 binds to downstream caspases, such as pro-caspase-9, and processes them into proteolytically active forms. This begins a caspase cascade resulting in apoptosis. In addition, Smac/Diablo is released from the mitochondria and blocks IAP proteins which normally interact with caspase-9 to inhibit apoptosis.")
26
Übersicht über die bekannten Apoptosemechanismen

27
Deregulierte Apoptose

28
Apoptose und AD Der neuronale Tod in degenerativen Erkrankungen ist selektiv und betrifft individuelle Zellen Es finden keine entzündlichen Prozesse statt Es kommt zur Aktivierung von Caspasen Es handelt sich nicht um eine offenkundige Apoptose. Wegen des chronischen Verlaufes der AD sind immer nur wenige apoptotische Neurone nachweisbar Selektivität: Apoptose findet hauptsächlich im Cortex und im limbischen System statt (in Parkinson z.B. im Hirnstamm und dort monoaminerge Neuronen) DNA – Fragmentierung war nicht überzeugend nachzuweisen Caspase 2 bzw 12 knock-out Mäuse resistent gegen Abeta Toxizität Wegen des chron. Verlaufes der AD sind immer nur wenige apoptot. Neurone nachweisbar Apoptose wahrscheinl. Nicht der einzige Mechanismus der Neurodegeneration
 DNA – Fragmentierung war nicht überzeugend nachzuweisen. Caspase 2 bzw 12 knock-out Mäuse resistent gegen Abeta Toxizität. Wegen des chron. Verlaufes der AD sind immer nur wenige apoptot. Neurone nachweisbar. Apoptose wahrscheinl. Nicht der einzige Mechanismus der Neurodegeneration.")
29
Bedeutung des Ab für die Apoptose in AD
Extrazelluläres Ab aktiviert Caspasen über den Rezeptorweg Ab akkumuliert auch intrazellulär im ER oder in Endosomen und kann den intrinsischen Weg der Apoptose auslösen Ab kann an die mitochondriale Alkoholdehydrogenase binden und die intrinsische Antwort auslösen Ab amplifiziert die Konsequenzen anderer Schädigungen (oxidativer Stress, Sauerstoff/Glucose-Entzug) Eine der Konsequenzen der durch Ab ausgelösten Apoptose ist die Spaltung des tau – Proteins Ab aktiviert p53 promoter Durch Ab wird die Spaltung des tau Proteins an Asp421 getriggered. Caspasen spalten aber auch an anderen Stellen. Abeta ist in vivo (Loo et al, nächste Folie) und in vitro neurotoxisch Abeta könnte Apoptose induzieren durch: Interaktion mit neuronalen Rezeptoren Aktivierung von caspasen Caspase2 und 12 defiziente Mäuse sind unempfindlich Möglicherweise rel. Lange Initiationsphase: Caspaseaktivität detektierbar ohne Zelltod chron. Caspaseaktivierung trägt zum pathologischenErscheinungsbild bei
 Eine der Konsequenzen der durch Ab ausgelösten Apoptose ist die Spaltung des tau – Proteins. Ab aktiviert p53 promoter. Durch Ab wird die Spaltung des tau Proteins an Asp421 getriggered. Caspasen spalten aber auch an anderen Stellen. Abeta ist in vivo (Loo et al, nächste Folie) und in vitro neurotoxisch. Abeta könnte Apoptose induzieren durch: Interaktion mit neuronalen Rezeptoren. Aktivierung von caspasen. Caspase2 und 12 defiziente Mäuse sind unempfindlich. Möglicherweise rel. Lange Initiationsphase: Caspaseaktivität detektierbar ohne Zelltod chron. Caspaseaktivierung trägt zum pathologischenErscheinungsbild bei.")
30
Bedeutung des Ab für die Apoptose in AD
RAGE – receptor for advanced glycation endproducts kann Radikalbildung induzieren P75 – neutrophin rezeptor induziert neuronalen Zelltod APP kann ebenfalls Apoptose induzieren Aktivierung von Mikroglia durch Abeta Freisetzung von Faktoren die Apoptose induzieren Abeta iduziert oxidativ. Stress und erhöhte cCa2+

31
Bedeutung des tau – Proteins für die Apoptose in AD
PHF Intrazelluläre Ablagerung Tau erfährt Konformationsänderungen in AD Es entstehen MC1- reaktive, gefaltete Strukturen des tau – Proteins Die MC1- Konformation ist spezifisch für tau in Fibrillenbündeln Phosphorylierung und Proteolyse ermöglichen die MC1 – Konformation Tau wird durch Calpain (Ca2+-abhängig) und Caspasen in ähnliche Fragmente gespalten Tau wirkt pro-apoptotisch Phosphoepitope, die im normalen tau nicht vorkommen In vitro ist Phosphorylierung nicht notwendig für die Fibrillenbildung, Proteolyse ist ausreichend Calpain arbeitet in Abhängigkeit von Ca2+; Calcium wird auch bei einer Reihe pathogenetischer Szenarios ausgeschüttet, die zu neurodegenerativen Erscheinungen führen. Ca wird aber auch bei der durch ER-Stress induzierten Apoptose verstärkt ausgeschüttet. Mögl Kinase u.a. cdk5
 und Caspasen in ähnliche Fragmente gespalten. Tau wirkt pro-apoptotisch. Phosphoepitope, die im normalen tau nicht vorkommen. In vitro ist Phosphorylierung nicht notwendig für die Fibrillenbildung, Proteolyse ist ausreichend. Calpain arbeitet in Abhängigkeit von Ca2+; Calcium wird auch bei einer Reihe pathogenetischer Szenarios ausgeschüttet, die zu neurodegenerativen Erscheinungen führen. Ca wird aber auch bei der durch ER-Stress induzierten Apoptose verstärkt ausgeschüttet. Mögl Kinase u.a. cdk5.")
32
Caspase-Spaltung von tau führt zur Ausbildung von NFT
Stimuli, die zur Caspasen aktivieren, führen zur Spaltung von Mikrotubuli-assoziiertem tau an der Aminosäure Asp421. Gespaltenes tau geht in die MC1- Konformation über. Die MC1-Konformation tendiert zu einer erhöhten Filamentbildung und zur Bildung von Aggregaten aus tau und Dtau. Die Aggregate behindern den Kinesin-vermittelten Transport entlang der Mikrotubuli. Die zelluläre Antwort auf die Blockade besteht u.U. in einer Hyperphosphorylierung, die zur Dissoziation von tau von den Mikrotubuli führt. Das hyperphosphorylierte tau bildet die paired helical filaments PHF Mutationen im Tau-Gen führen zwar zu NFTs, aber nicht zu senilen Plaques, aber umgekehrt APP- oder PS- Mutation führt zu Plaques und NFTs

33
Zusammenfassung der molekularen Vorgänge
bei der Apoptose in AD

34
Zusammenfassung der molekularen Vorgänge
bei der Apoptose in AD

35
Therapeutische Intervention in die apoptotischen Prozesse kann die Progression der Alzheimer – Demenz verlangsamen

Ähnliche Präsentationen












>")






