Präsentation herunterladen
Die Präsentation wird geladen. Bitte warten

1
“Inner-sphere electron transfer“: IUPAC
Historically an electron transfer between two metal centres sharing a ligand or atom in their respective coordination shells. The definition has more recently been extended to any situation in which the interaction between the donor- and acceptor centres in the transition state is significant (>20 kJ/mol)
")
3
vorgelagertes Brückenbildungsgleichgewicht
Oft erfolgt der Vorgang der Brückenbildung schnell und reversibel vorgelagertes Brückenbildungsgleichgewicht schnell langsam schnell Geschwindigkeitsbestimmender Schritt: Elektronenübertragung auf der Brücke Es ergibt sich eine zusammengesetzte Geschwindigkeitskonstante 2. Ordnung (fast immer der Fall).
.")
4
Reaktionsgeschwindigkeit hängt von der Art der Brücke ab (von ihrer Wirksamkeit als Vermittler des Elektronentransfers) Quelle: Tobe

5
Es gibt Systeme, in denen mehrfache Brücken gebildet und sogar übertragen werden, wie z.B. die Reaktion zwischen cis-Tetraaqua-diazido-Chrom (III) und Hexaaqua-Cr(II).
 und Hexaaqua-Cr(II)..")
6
Oxidations- und Reduktionsmittel ändern ihre Oxidationsstufen um die gleiche Zahl von Einheiten.
Oxidations- und Reduktionsmittel ändern ihre Oxidationsstufen um eine unterschiedliche Zahl von Einheiten. Dies erfolgt nie in einem Schritt: Zumindest eine der Komponenten muss eine instabile Oxidationsstufe durchlaufen.

7
Nichtkomplementäre Redoxreaktionen
Zur Aufklärung des Mechanismus ist es nötig, die reaktive Zwischenverbindung (enthält die instabile Oxidationsstufe) zu identifizieren. Frage: Wieviele e- werden gleichzeitig übertragen? Allgemein gilt: Der Bildungsprozess der instabilen Zwischenverbindung ist reversibel, es stellt sich ein vorgelagertes Gleichgewicht ein, dem ein zweiter Reaktionsschritt folgt.
 zu identifizieren. Frage: Wieviele e- werden gleichzeitig übertragen Allgemein gilt: Der Bildungsprozess der instabilen Zwischenverbindung ist reversibel, es stellt sich ein vorgelagertes Gleichgewicht ein, dem ein zweiter Reaktionsschritt folgt.")
8
Zwei denkbare Reaktionswege a) oder b), welcher trifft zu? a)
Folge von Einelektronenübergängen, instabile Zwischenverbindung ist Tl(II). b) Zugabe von Fe(III) verlangsamt die Reaktion, Zugabe von Tl(I) aber nicht. Zweielektronenübergang, gefolgt von Einelektronenübergang, instabile Zwischenverbindung ist Fe(IV).
. b) Zugabe von Fe(III) verlangsamt die Reaktion, Zugabe von Tl(I) aber nicht. Zweielektronenübergang, gefolgt von Einelektronenübergang, instabile Zwischenverbindung ist Fe(IV).")
9
Nichtkomplementäre Redoxreaktionen sind gewöhnlich langsam, weil eine der Komponenten gezwungen ist, eine ungewöhnliche Oxidationsstufe anzunehmen: Diese ist energetisch ungünstig, und ihr Bildungsprozess ist reversibel, Zwischenverbindung liegt in nur geringer Konzentration vor Langsame Reaktion Wenn durch einen Katalysator die ansonsten notwendige Bildung einer instabilen Oxidationsstufe vermieden wird, kann das die Reaktion sehr beschleunigen! Daher sind nichtkomplementäre Redoxreaktionen stark von Verunreinigungen durch Metallionen abhängig.

10
Oxidation von Cr(III) zu Cr(VI) durch Peroxodisulfat erfolgt sehr langsam.
Peroxodisulfat ist ein sehr starkes Oxidationsmittel, aber es wirkt als Zweielektronen-Oxidationsmittel. Oxidation von Spezies, die Einelektronenübergänge bevorzugen, erfolgt langsam. AgNO3 als Katalysator

11
Ag(II) betätigt sich dann als Einelektronen-Oxidationsmittel:
Quantitative Bestimmung von Chrom(III)
")
12
Photochemische Redoxreaktionen
Man kann einer Reaktion auf unterschiedliche Art Aktivierungsenergie zuführen: thermische Energie durch Erhöhung der Temperatur. „statistisch breite“ Methode Breite Streuung individueller molekularer Energien. Strahlungsenergie Photochemische Anregung, kann viel präziser sein. Allerdings muss das Molekül imstande sein, das Lichtquant auch zu absorbieren, d.h. es muss einen erreichbaren angeregten Zustand geben, der sich im richtigen energetischen Abstand zum Grundzustand befindet.

13
Photochemische Reaktionen: Allgemeine Definition
Unter photochemischen Reaktionen versteht man Reaktionen, die nicht ausschließlich (wie es bei thermischen Reaktionen der Fall ist) im elektronischen Grundzustand ablaufen Sondern bei denen entlang der Reaktionskoordinate auch elektronisch angeregte Zustände involviert sind.
 im elektronischen Grundzustand ablaufen. Sondern bei denen entlang der Reaktionskoordinate auch elektronisch angeregte Zustände involviert sind.")
14
Vereinfachtes Orbitaldiagramm eines oktaedrischen Übergangsmetallkomplexes mit verschiedenen elektronischen Übergängen

15
Substitutions- und Isomerisierungsreaktionen aus angeregten Zuständen
π π* Übergänge im Liganden und auch d-d Übergänge im Metall (bzw. Metallion) führen nicht zu einer Änderung der Oxidationszahl des Metalls, nur zu einer Änderung der Elektronendichteverteilung. Übergänge von bindenden (bei Metallen auch nicht bindenden) in antibindende Zustände schwächen die Metall-Ligand Bindung. Damit treten Substitutions- und Isomerisierungsreaktionen auf.
 führen nicht zu einer Änderung der Oxidationszahl des Metalls, nur zu einer Änderung der Elektronendichteverteilung. Übergänge von bindenden (bei Metallen auch nicht bindenden) in antibindende Zustände schwächen die Metall-Ligand Bindung. Damit treten Substitutions- und Isomerisierungsreaktionen auf.")
16
Die Selektivität der photochemischen Anregung kann dazu verwendet werden, darüber zu bestimmen, welches Produkt entsteht: d6 Co3+ Komplex (high spin) Absorptionspeak für grünes Licht: d-d Übergang Absorptionspeak für UV: Ligand zu Metall charge-transfer Übergang (e- wechselt vom N3- zum Co3+) Bestrahlt man mit UV, so entstehen Co(II) und ·N3 (2 N N2) Durch Änderung der Frequenz kann man statt einer Ligandensubstitutions-Reaktion eine Redoxreaktion erhalten.
 Absorptionspeak für grünes Licht: d-d Übergang. Absorptionspeak für UV: Ligand zu Metall charge-transfer Übergang (e- wechselt vom N3- zum Co3+) Bestrahlt man mit UV, so entstehen Co(II) und ·N3 (2 N3 3 N2) Durch Änderung der Frequenz kann man statt einer Ligandensubstitutions-Reaktion eine Redoxreaktion erhalten.")
17
Photochemische Einelektronenanregung führt zu einer Spezies, die sowohl ein besseres Oxidationsmittel als auch ein besseres Reduktionsmittel ist als die ursprüngliche nicht angeregte Spezies. Besseres Oxidationsmittel, weil leeres Orbital niedriger Energie vorhanden. Besseres Reduktionsmittel, weil hochenergetisches antibindendes Elektron vorhanden. Quelle: Porterfield

18
Redox-Photochemie von Co3+ Komplexen (d6)
Co(III) Komplexe erfahren charakteristischerweise LMCT-Übergänge (ligand-to-metal charge transfer) Dabei entstehen gegen Substitution labile Co2+ Komplexe Photoredoxreaktion oft von Substitution begleitet
 Komplexe erfahren charakteristischerweise. LMCT-Übergänge. (ligand-to-metal charge transfer) Dabei entstehen gegen Substitution labile Co2+ Komplexe. Photoredoxreaktion oft von Substitution begleitet.")
20
diphos=bis-Diphenylphosphinoethan
+ + H L4Ir - H L4Ir + L4Ir + H2 + Hier werden keine freien H Radikale erzeugt, die extrem starke Base H- kann das Proton aus der anderen Ir-H Bindung abspalten.

21
Die Spezies Ru(bipy)32+* ist der angeregte Zustand, der aus einem MLCT Übergang hervorgeht
Obwohl der Ru2+ Komplex in wässriger Lösung gegen Disproportionierung stabil ist, disproportioniert der angeregte Zustand spontan zu der stark oxidierenden (3+) - Spezies und der stark reduzierenden (1+) -Spezies.
 - Spezies und der stark reduzierenden (1+) -Spezies.")
22
Man kann den angeregten Zustand (Triplett-Zustand) Ru(bipy)32+
Man kann den angeregten Zustand (Triplett-Zustand) Ru(bipy)32+* auch chemisch herstellen, indem man Ru(bipy)33+ in wässriger Lösung mit N2H4 reduziert. Es entsteht zunächst Ru(bipy)32+* Bei der Rückkehr zum (2+)-Grundzustand wird ein orangefarbenes Licht emittiert (Chemilumineszenz bei λ=610 nm). aus: D.F. Shriver and P.W. Atkins, Inorganic Chemistry
 Ru(bipy)32+* auch chemisch herstellen, indem man Ru(bipy)33+ in wässriger Lösung mit N2H4 reduziert. Es entsteht zunächst Ru(bipy)32+* Bei der Rückkehr zum (2+)-Grundzustand wird ein orangefarbenes Licht emittiert (Chemilumineszenz bei λ=610 nm). aus: D.F. Shriver and P.W. Atkins, Inorganic Chemistry.")
23
Quelle: Wöhrle et al.

24
aus: Wöhrle/Tausch/Stohrer Photochemie

25
Substitutionsreaktionen bei Übergangsmetallkomplexen
Substitution=Ersatz eines Liganden aus der Koordinationsschale durch einen anderen aus deren Umgebung. Die Koordinationszahl ist vor und nach der Reaktion gleich, ändert sich nur kurzzeitig während der Reaktion. Molekularität der nucleophilen Substitution: Man spricht von SN1 (monomolekularen) und SN2 (bimolekularen) Reaktionen. Die Molekularität kann definiert werden in Abhängigkeit vom zeitlichen Verlauf der Bindungsbildung und Bindungsspaltung bei der Substitution.
 und SN2 (bimolekularen) Reaktionen. Die Molekularität kann definiert werden in Abhängigkeit vom zeitlichen Verlauf der Bindungsbildung und Bindungsspaltung bei der Substitution.")
26
Bindungsbildung und –Spaltung können synchron oder asynchron verlaufen
Ein synchroner Prozess verläuft in einem Schritt und weist einen ÜZ, aber keine Zwischenverbindung auf. Der ÜZ bestimmt die energetischen Verhältnisse (Reaktionsgeschwindigkeit) und den stereochemischen Verlauf der Reaktion Beim asynchronen Prozess gibt es zwei Möglichkeiten: a) zuerst Bindungsspaltung, dann Bindungsbildung b) zuerst Bindungsbildung, dann Bindungsspaltung In beiden Fällen a) und b) tritt eine Zwischenverbindung auf!
 und den stereochemischen Verlauf der Reaktion. Beim asynchronen Prozess gibt es zwei Möglichkeiten: a) zuerst Bindungsspaltung, dann Bindungsbildung. b) zuerst Bindungsbildung, dann Bindungsspaltung. In beiden Fällen a) und b) tritt eine Zwischenverbindung auf!")
27
Wenn die Bindungsspaltung zuerst erfolgt, hat die Zwischenverbindung eine niedrigere Koordinationszahl, der Vorgang wird als dissoziativ bezeichnet Wenn die Bindungsbildung zuerst erfolgt, hat die Zwischenverbindung eine höhere Koordinationszahl , der Prozess wird als assoziativ bezeichnet

29
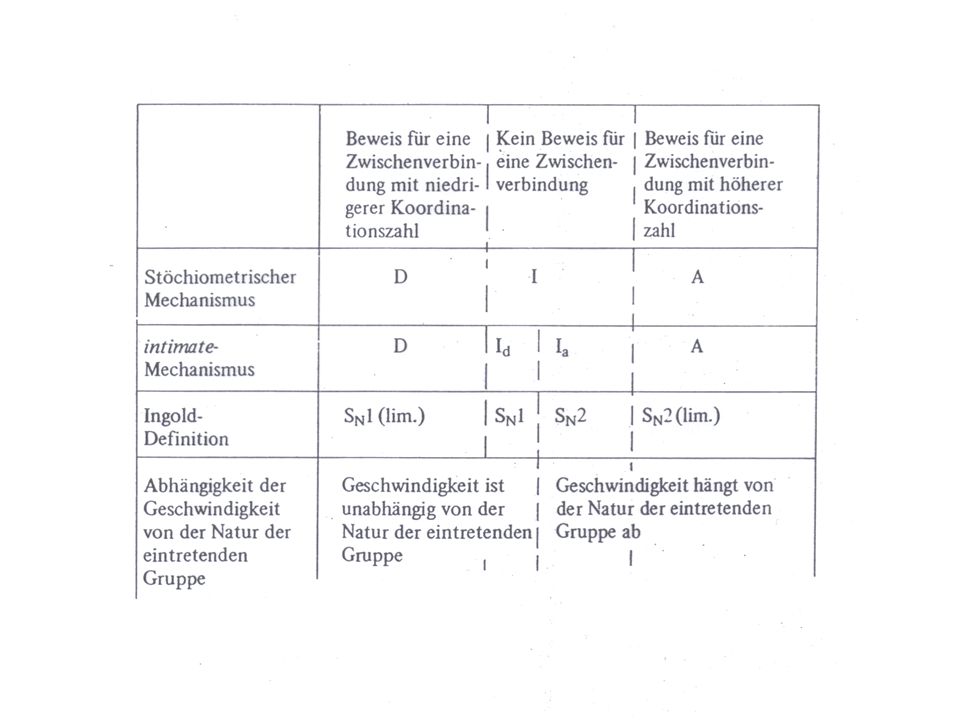
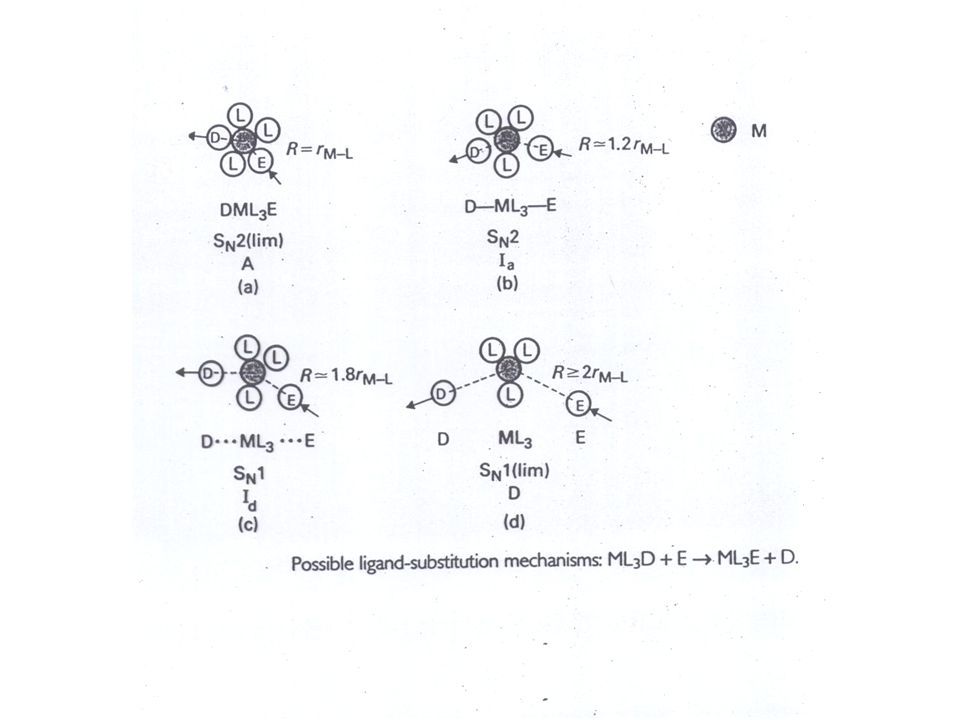
Definition nach Langford und Gray
D= dissoziativer Prozess A= assoziativer Prozess I=synchroner Prozess (interchange) Id = (dissoziativ) im ÜZ tritt die eintretende Gruppe nicht direkt mit dem Reaktionszentrum in Wechselwirkung Ia=(assoziativ) im ÜZ erfolgt Bindungsbildung zwischen der eintretenden Gruppe und dem Reaktionszentrum asynchron Id Ia intimate = engerer Mechanismus
 Id = (dissoziativ) im ÜZ tritt die eintretende Gruppe nicht direkt mit dem Reaktionszentrum in Wechselwirkung. Ia=(assoziativ) im ÜZ erfolgt Bindungsbildung zwischen der eintretenden Gruppe und dem Reaktionszentrum. asynchron. Id Ia. intimate = engerer Mechanismus.")
31
Das Übergangsmetallatom oder –Ion mit seiner inneren und äußeren Koordinationssphäre:
Die Liganden der inneren Koordinationssphäre sind jeweils durch ein gemeinsames Elektronenpaar koordinativ gebunden Die äußere Koordinationssphäre besteht aus potentielle Liganden, die nur durch Dipol-Wechselwirkungen oder Ionen-Dipol Wechselwirkungen schwach festgehalten werden Metall – Ligand Abstand rM-L Wir betrachten den Augenblick, wenn der eintretende Ligand E (entering) und der austretende Ligand D (departing) den genau gleichen Abstand vom Metallatom M haben rM-D=rM-E
 und der austretende Ligand D (departing) den genau gleichen Abstand vom Metallatom M haben rM-D=rM-E.")
32
Wenn sich E in der äußeren Koordinationssphäre befindet und D in der inneren Koordinationssphäre, dann ist

34
Der Zusammenhang zwischen der kinetischen Reaktionsordnung und dem Reaktionsmechanismus ist oft dadurch verkompliziert, dass das Lösungsmittel selbst als Nucleophil wirkt. enormer Konzentrationsvorteil gegenüber anderen nucleophilen Liganden in Lösung! Reaktion 1 Oft misst man, so glaubt man, die Reaktionsgeschwindigkeit von Reaktion 1. Statt dessen misst man die RG von Reaktion 2: Reaktion 2 pseudo-1.Ordnung langsamer als Reaktion 3 Reaktion 3 Anation (=Umkehrung der Solvolyse)
")
35
Folge von Hydrolyse und Anation

36
Ligandensubstitution in oktaedrischen Komplexen
Kinetische Studien an oktaedrischen Komplexen in saurer wässriger Lösung zeigen in den meisten Fällen ein Geschwindigkeitsgesetz 1. Ordnung und keinen Einfluss des eintretenden Liganden auf die Reaktionsgeschwindigkeit Weist dies auf einen dissoziativen Mechanismus hin?

38
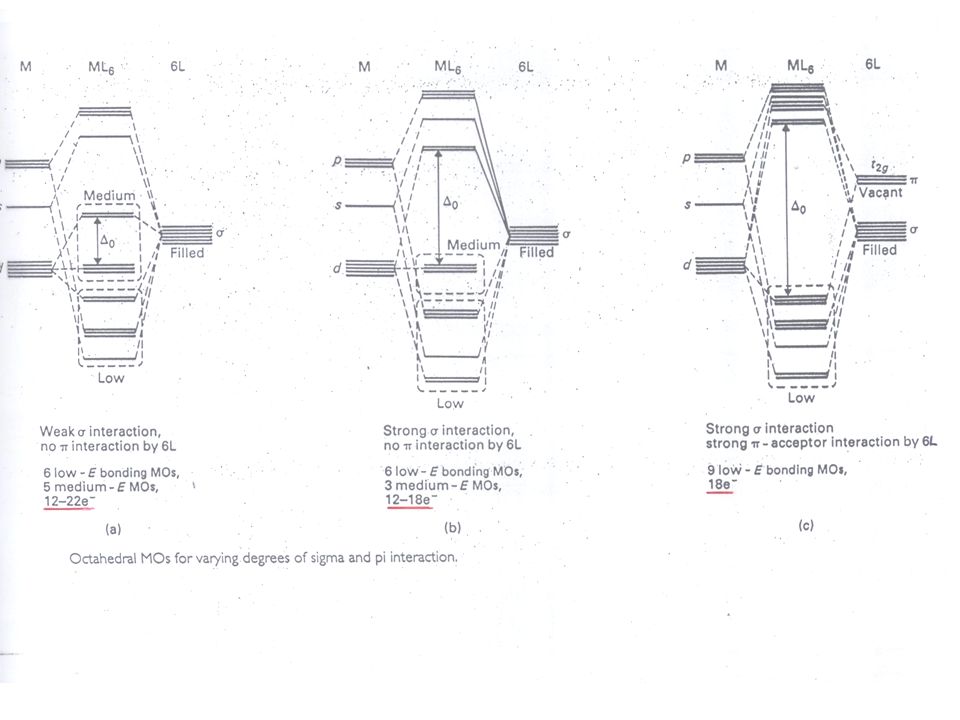
Sterische Gründe „Sterische Absättigung“ bei Koordinationszahl Z=6
Z>6 benötigt eine erhöhte Aktivierungsenergie, dies führt zu einer nicht konkurrenzfähigen langsamen Reaktionsgeschwindigkeit im Falle eines Reaktionswegs über einen siebenfach koordinierten ÜZ Für Co3+ Komplexe gilt speziell, dass wenn 6 Liganden 6 Elektronenpaare beisteuern, gerade 18 Valenzelektronen vorhanden sind Ein weiterer Ligand in einem ÜZ mit erhöhter Koordinationszahl würde eine sehr ungünstige elektronische Energie für sein Elektronenpaar vorfinden, siehe oktaedrische Molekülorbitale

40
Geringer Einfluss der eintretenden Gruppe E auf die Reaktionsgeschwindigkeitskonstante
Zu erwarten, wenn die Dissoziation der geschwindigkeitsbestimmende Schritt des Mechanismus ist. Beispiel: Die Geschwindigkeitskonstanten der Reaktionen von [Co(NH3)5(H2O)]3+ mit Cl-, Br-, NCS-, N3-, NO3-, H2PO4-, NH3 liegen alle sehr nahe beisammen mit k=1.3x10-6 bis k=2.5x10-6 L mol-1 s-1 (25°C) . Wäre der Prozess assoziativ, dann müssten die Bindungseigenschaften des hereinkommenden Liganden sehr stark die Geschwindigkeitskonstante beeinflussen!
5(H2O)]3+ mit Cl-, Br-, NCS-, N3-, NO3-, H2PO4-, NH3 liegen alle sehr nahe beisammen mit k=1.3x10-6 bis k=2.5x10-6 L mol-1 s-1 (25°C) . Wäre der Prozess assoziativ, dann müssten die Bindungseigenschaften des hereinkommenden Liganden sehr stark die Geschwindigkeitskonstante beeinflussen!")
41
Großer Einfluss der austretenden Gruppe D auf die Geschwindigkeitskonstante
Dann zu erwarten, wenn der Bruch der M-D Bindung der geschwindigkeitsbestimmende Schritt ist. Kinetische Daten für die saure Hydrolyse von [Co(NH3)5X]2+ zeigen, dass die Geschwindigkeitskonstanten für verschiedene Liganden X über 8 Größenordnungen variieren!
5X]2+ zeigen, dass die Geschwindigkeitskonstanten für verschiedene Liganden X über 8 Größenordnungen variieren!")
43
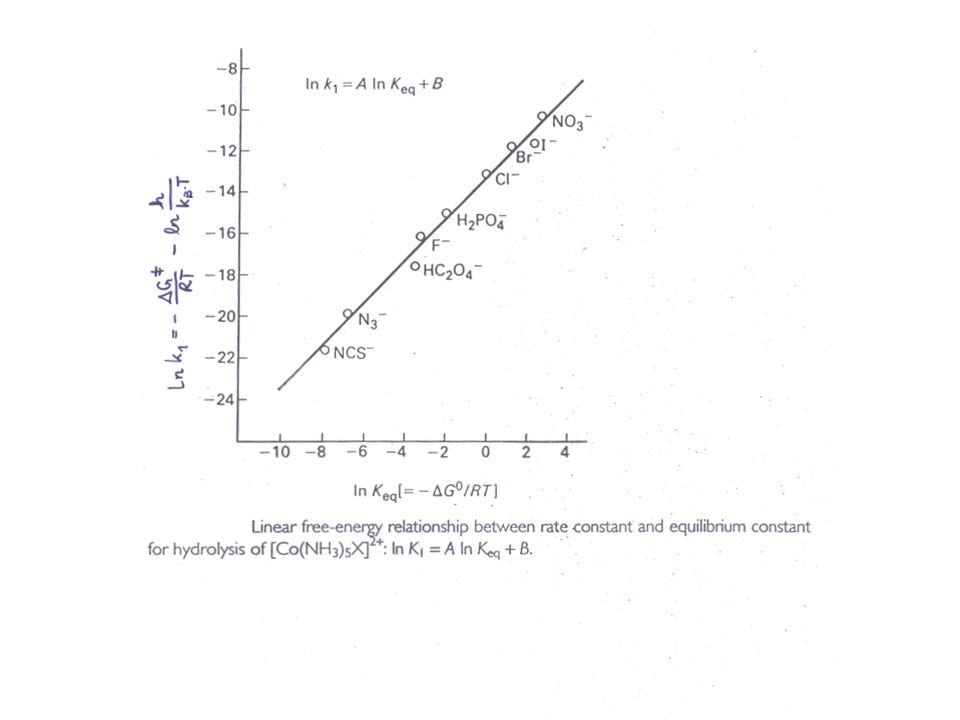
Zusammenhang zwischen Gleichgewichtskonstante und Geschwindigkeitskonstante
saure Hydrolyse von [Co(NH3)5X]2+ ln k1 aufgetragen gegen ln K für verschiedene Liganden X ergibt eine Gerade, mit dem Anstieg 1.03 d.h und haben fast den gleichen Wert!
5X]2+ ln k1 aufgetragen gegen ln K für verschiedene Liganden X ergibt eine Gerade, mit dem Anstieg d.h. und haben fast den gleichen Wert!")
46
ÜZ ähnlich dem Reaktionsprodukt!
D.h. beide haben ähnliche Energie und Struktur Der ÜZ muss also eine Konfiguration des Systems sein, bei der sich das X- Ion bereits vom Co3+ abgetrennt hat Dissoziativer Mechanismus Der Unterschied zwischen ÜZ und Produkt ist nur, dass das H2O Molekül noch in der äußeren Koordinationssphäre liegt

Ähnliche Präsentationen




















![Für einen inner-sphere Elektronentransfer ist ein Ligandentransfer nicht unbedingt nötig, z.B. Oxidation von [Cr(H2O)6]2+ mit [IrCl6]2- [IrCl6]2- + [Cr(H2O)6]2+](/3/891079/big_thumb.jpg "Für einen inner-sphere Elektronentransfer ist ein Ligandentransfer nicht unbedingt nötig, z.B. Oxidation von [Cr(H2O)6]2+ mit [IrCl6]2- [IrCl6]2- + [Cr(H2O)6]2+>")


>")
