Präsentation herunterladen
Die Präsentation wird geladen. Bitte warten

1
Interpretation von Geschwindigkeitskonstanten nahe der diffusionskontrollierten Grenze
Zweistufiges Schema: 1. Diffusion der reagierenden Teilchen zueinander und voneinander weg 2. Reaktion innerhalb des Lösungsmittelkäfigs LM-Käfig

2
Steady-state approximation:

3
Für sehr großes kr gilt: kexp →kdc

4
Ionenreaktionen in Lösungen
Die elektrostatischen Kräfte zwischen Ionen beeinflussen bestimmte Eigenschaften wie Aktivitätskoeffizienten und elektr. Leitfähigkeit sowie die Geschwindigkeitskonstanten bei Ionenreaktionen Auch die Dielektrizitätskonstante (=dielektrische Leitfähigkeit) des LM spielt eine wichtige Rolle, da mit abnehmendem ε die elektrostatischen Kräfte zwischen den Ionen zunehmen.
 des LM spielt eine wichtige Rolle, da mit abnehmendem ε die elektrostatischen Kräfte zwischen den Ionen zunehmen.")
5
Einfluss der Dielektrizitätskonstante ε des Lösungsmittels
Zwei Ionen befinden sich im Abstand r zueinander. Die elektrostatische Kraft zwischen diesen Ionen ist (Coulomb‘sches Gesetz): Um diesen Abstand um die Strecke -dr zu vermindern, müssen wir die folgende Arbeit aufwenden: = dielektrische Leitfähigkeit oder Dielektrizitätskonstante
: Um diesen Abstand um die Strecke -dr zu vermindern, müssen wir die folgende Arbeit aufwenden: = dielektrische Leitfähigkeit oder Dielektrizitätskonstante.")
6
Um zwei Ionen aus unendlicher Entfernung auf ihren Stoßdurchmesser dAB zu bringen, muss folgender Betrag an (elektrostatischer) Arbeit aufgebracht werden: w zählt mit zum Arbeitsaufwand bei der Bildung des Aktivierten Komplexes! positiv, wenn zA und zB gleiches Vorzeichen haben: Aktivierungsenergie erhöht negativ, wenn zA und zB ungleiches Vorzeichen haben: Aktivierungsenergie verringert

7
elektrostatisch nicht-elektrostatisch

8
ln k ist eine lineare Funktion von 1/ε
Erst bei kleinen Werten von ε tritt Abweichung von der Geraden auf (durch Ionenassoziation)
")
9
Einfluss gelöster Salze
Beispiel: Bimolekulare Reaktion Die beiden Ionen AzA und BzB reagieren miteinander Die Reaktion verläuft über den aktivierten Komplex (AB)zA+zB AzA + BzB (AB)zA+zB P z.B. Fe3+ + I (FeI) Fe2+ + ½ I2
zA+zB. AzA + BzB (AB)zA+zB P. z.B. Fe3+ + I- (FeI)2+ Fe2+ + ½ I2.")
10
Da Ionen vorliegen, muss die Quasi Gleichgewichtskonstante K
Da Ionen vorliegen, muss die Quasi Gleichgewichtskonstante K* durch Aktivitäten ausgedrückt werden: In die Reaktionsgeschwindigkeit geht jedoch die Konzentration des aktivierten Komplexes ein, nicht seine Aktivität!

12
Nach der Debye-Hückel-Theorie gilt für wässrige Lösungen bei 298 K (Debye-Hückelsches Grenzgesetz für verdünnte Lösungen) Die Summierung erstreckt sich über alle Ionenarten in der Lösung, nicht nur über die reagierenden Ionen!

13
Brønsted‘sche Gleichung

14
Trägt man für eine wässrige Lösung bei 298 K gegen
Die Quadratwurzel der Ionenstärke auf, so erhält man eine Gerade, deren Steigung nahezu gleich dem Produkt der Ionenladungen der reagierenden Ionen ist.

15
Änderung der Reaktionsgeschwindigkeitskonstante mit der Ionenstärke = Primärer kinetischer Salzeffekt Wenn zA und zB dasselbe Vorzeichen haben, dann ist die Steigung der Geraden positiv, die Reaktionsgeschwindigkeit nimmt mit steigender Ionenstärke zu Wenn zA und zB unterschiedliches Vorzeichen haben, dann ist die Steigung der Geraden negativ, die Reaktionsgeschwindigkeit nimmt mit steigender Ionenstärke ab Ist einer der Reaktionsteilnehmer ungeladen, dann ist die Reaktionsgeschwindigkeit unabhängig von der Ionenstärke

16
Quelle: Moore/Hummel

17
Rohrzuckerinversion Die Saccharose hydrolisiert in wässriger Lösung protonenkatalysiert zu D-(+)- Glucose und D-(-)-Fructose. Die Sauerstoffbrücke wird in einem vorgelagerten Gleichgewicht protoniert und schließlich hydrolytisch gespalten: SH+ + H2O → G + F + H+ Einer der Reaktionsteilnehmer (H2O) ist ungeladen.
- Glucose und D-(-)-Fructose. Die Sauerstoffbrücke wird in einem vorgelagerten Gleichgewicht protoniert und schließlich hydrolytisch gespalten: SH+ + H2O → G + F + H+ Einer der Reaktionsteilnehmer (H2O) ist ungeladen.")
18
Kontrolle der experimentellen Bedingungen
Enthält das System nur die reagierenden Ionen, so ändert sich während der Reaktion die Ionenstärke oft beträchtlich. Dementsprechend ändert sich der Wert der Geschwindigkeitskonstante während der Reaktion. Um zuverlässige Werte für die Geschwindigkeitskonstanten von Ionenreaktionen zu bekommen, setzt man der Reaktionsmischung einen Überschuss eines inerten Salzes zu (schwach koordinierende Ionen wie NaNO3 oder NaClO4 oder Natrium-Trifluoromethansulfonat) Dadurch bleibt die Ionenstärke während der Reaktion praktisch konstant.
 Dadurch bleibt die Ionenstärke während der Reaktion praktisch konstant.")
19
Geltungsbereich der Brønsted‘schen Gleichung:
Nur bei Konzentrationen erfüllt, die auch im Geltungsbereich der Debye-Hückel Theorie liegen. Bei höheren Konzentrationen treten Ionenassoziationen auf. Es ist dann nicht mehr möglich, alle Salzeffekte in dem einfachen Faktor für die Ionenstärke zusammenzufassen!

20
Kinetische Isotopieeffekte
Können sehr hilfreich sein, wenn die Frage auftritt, ob während des Aktivierungsprozesses eine Bindung zu Wasserstoff oder einem anderen leichten Element gebrochen wird oder nicht. Betrachten wir eine Bindung zwischen einer Gruppe R und einem Atom A. Während der Reaktion erfolge der Bruch dieser Bindung. Wie unterscheidet sich die Geschwindigkeitskonstante dieser Reaktion von der einer analogen Reaktion, bei der A durch A* (= schwereres Isotop desselben Elements) ersetzt ist?
 ersetzt ist")
21
Die Chemische Bindung, betrachtet als quantenmechanischer harmonischer Oszillator
Harmonischer Oszillator bedeutet: Rückstellkraft proportional zur Auslenkung Beschleunigung proportional zur Auslenkung und dieser entgegengesetzt Die Energieniveaus des Oszillators sind: Schwingungsquantenzahl

22
Das niedrigste Schwingungsniveau liegt bei n=0 Nullpunktsenergie
Anders als in der klassischen Mechanik kann der Oszillator seine Energie nie ganz abgeben Das niedrigste Schwingungsniveau liegt bei n=0 Nullpunktsenergie Harmonischer Oszillator: Kreisfrequenz Auslenkung Amplitude

23
Kraftkonstante (Federkonstante)
reduzierte Masse Das Spektrum des harmonischen Oszillators besteht nur aus einer einzigen Frequenz, Diese ist (wie in der klassischen Mechanik) unabhängig von der Amplitude (Energie) der Schwingung Der Auslenkung aus der Ruhelage r – r0 wirkt die harmonische Kraft F entgegen: F = - k‘ (r – r0)
 unabhängig von der Amplitude (Energie) der Schwingung. Der Auslenkung aus der Ruhelage r – r0 wirkt die harmonische Kraft F entgegen: F = - k‘ (r – r0)")
24
Die Kraftkonstante k‘ ist unabhängig vom Isotop, daher gilt:
A sei H (Wasserstoff) A* sei D (Deuterium) Die Kraftkonstante k‘ ist unabhängig vom Isotop, daher gilt: mR>>mH mR>>mD
 A* sei D (Deuterium) Die Kraftkonstante k‘ ist unabhängig vom Isotop, daher gilt: mR>>mH. mR>>mD.")
25
Der Unterschied in den Aktivierungsenergien kann gleichgesetzt werden dem Unterschied zwischen den Grundzuständen, wenn im ÜZ die R-H bzw. R-D Bindung nicht mehr existiert und daher die Übergangszustände die gleiche Energie haben

27
Für eine C-H Bindung (aliphatisch) ist die Streck-Schwingungsfrequenz 8.7x1013 s-1
Bei 298 K ergibt sich daher Primärer kinetischer Isotopieeffekt: Wenn die Bindung zum Isotop gespalten wird Für H/D ist der Effekt am größten, für C-12/C-13 beträgt er nur ca. 4%

29
Hinweise auf die Struktur des Übergangszustandes
Wird die R-H bzw. R-D Bindung im ÜZ nicht ganz sondern nur teilweise gebrochen, so ist der kinetische Isotopieeffekt kleiner als berechnet. Der Effekt ist auch dann kleiner, wenn sich im ÜZ eine neue Bindung von H (bzw. D) zu einer zweiten an der Reaktion teilnehmenden Spezies abzuzeichnen beginnt. Ist die neue Bindung viel stärker als die alte, so kann man sogar einen inversen kinetischen Isotopieeffekt beobachten!
 zu einer zweiten an der Reaktion teilnehmenden Spezies abzuzeichnen beginnt. Ist die neue Bindung viel stärker als die alte, so kann man sogar einen inversen kinetischen Isotopieeffekt beobachten!")
30
Regel: Substitution durch ein schwereres Isotop favorisiert die Spezies mit der stärkeren Bindung in jedem Gleichgewicht, einschließlich des Quasi-Gleichgewichts des ÜZ mit den Edukten. Bindung R-H bzw. R-D stärker im Edukt als im ÜZ: Reaktion durch Ersatz von H durch D verlangsamt Bindung R-H bzw. R-D stärker im ÜZ als im Edukt: Reaktion durch Ersatz von H durch D beschleunigt (weil höhere Konzentration an aktiviertem Komplex)
")
31
Kinetik des Isotopenaustausches
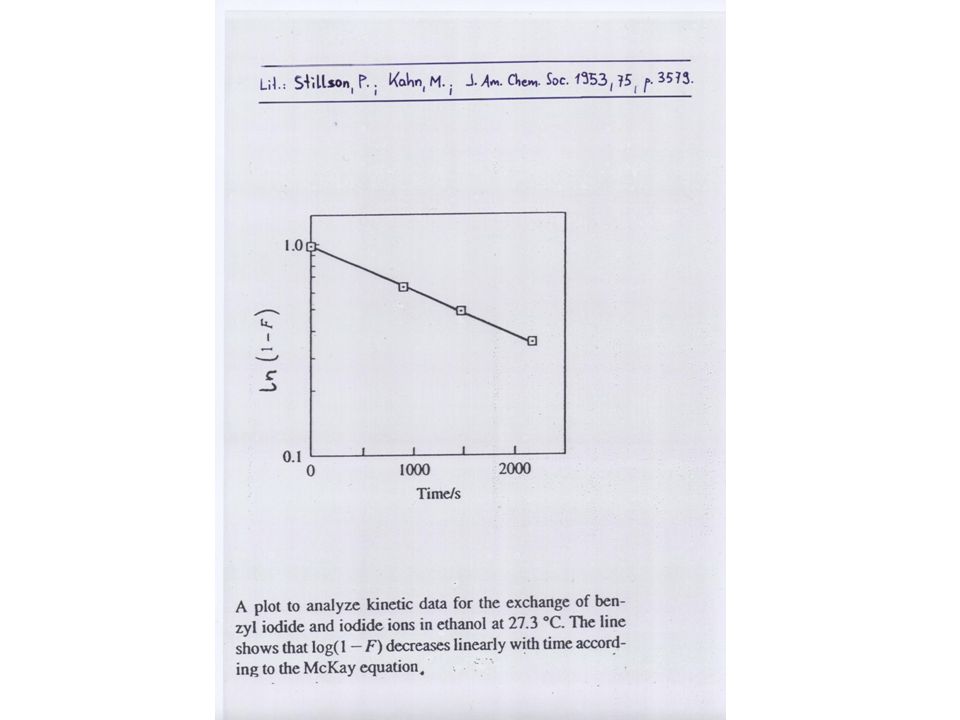
Reaktion AX + B*X A*X + BX *X ist z.B. ein radioaktives Isotop (Tracer) “self-exchange reaction“ Chemisches Gleichgewicht sei vorhanden, aber nicht Isotopengleichgewicht. Man nennt eine solche Reaktion Austauschreaktion. Findet auf alle Fälle statt, mit und ohne Markierungs-Isotop! Das Markierungs-Isotop macht den Austauschvorgang beobachtbar.
 self-exchange reaction Chemisches Gleichgewicht sei vorhanden, aber nicht Isotopengleichgewicht. Man nennt eine solche Reaktion Austauschreaktion. Findet auf alle Fälle statt, mit und ohne Markierungs-Isotop! Das Markierungs-Isotop macht den Austauschvorgang beobachtbar.")
32
(CH3)2CO + H2*O = (CH3)2C*O + H2O
Beispiele: (CH3)2CO + H2*O = (CH3)2C*O + H2O Cr(H2O)63+ + *Cr(H2O)62+ = Cr(H2O)62+ + *Cr(H2O)63+ (hier wird ein Elektron ausgetauscht) *O=18O (stabiles Isotop) *Cr=51Cr (Elektroneneinfang, t1/2=27.8 d) Es werden in bestimmten Zeitabständen Proben entnommen, nach geeigneten Trennungsschritten (Extraktion, Chromatographie) wird der Isotopengehalt z.B. mittels Massenspektrometrie oder Gamma-Spektrometrie bestimmt
2CO + H2*O = (CH3)2C*O + H2O. Cr(H2O)63+ + *Cr(H2O)62+ = Cr(H2O)62+ + *Cr(H2O)63+ (hier wird ein Elektron ausgetauscht) *O=18O (stabiles Isotop) *Cr=51Cr (Elektroneneinfang, t1/2=27.8 d) Es werden in bestimmten Zeitabständen Proben entnommen, nach geeigneten Trennungsschritten (Extraktion, Chromatographie) wird der Isotopengehalt z.B. mittels Massenspektrometrie oder Gamma-Spektrometrie bestimmt.")
Ähnliche Präsentationen
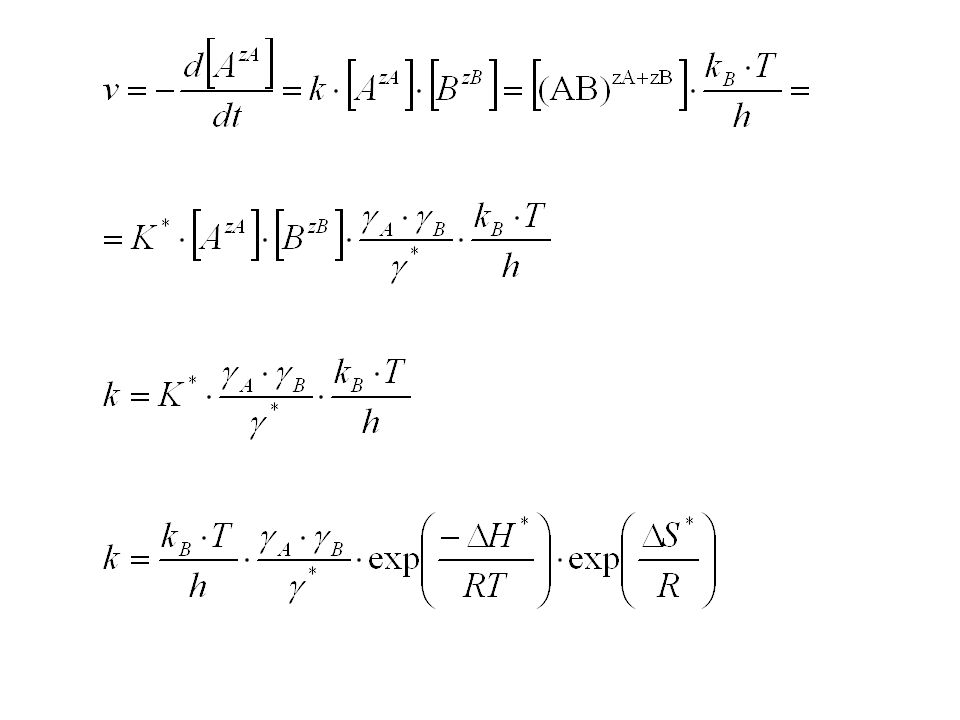
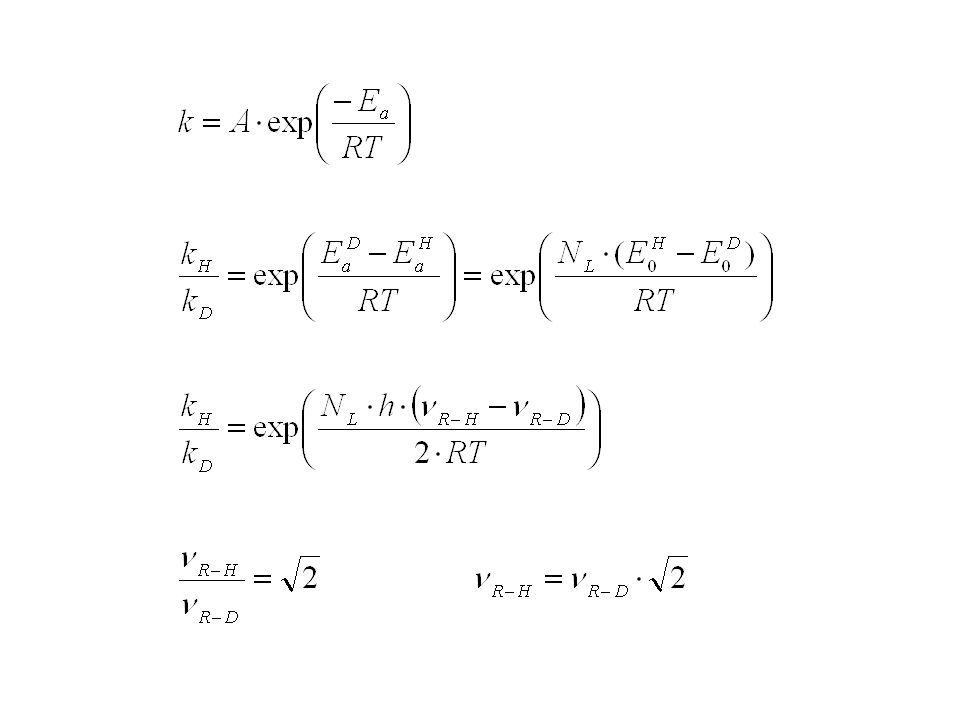









 Schwingungen>")













