Präsentation herunterladen
Die Präsentation wird geladen. Bitte warten

1
Austrian Agency for Health and Food Safetywww.ages.at Dr. Isabella Berger Mag. Pharm. Sonja Ernsthofer Institut Überwachung AGES Gespräch Wien, 29. Oktober 2015 Handhabung von Blutproben und Prüfmedikation

2
2 Handhabung von Blutproben Auszug aus dem Protokoll: PK Proben sind maximal 60 Minuten auf Eis zu lagern, anschließend für 10 Minuten bei 2-8°C bei 1900xg zu zentrifugieren und das dekantierte Plasma anschließend innerhalb von 90 Minuten ab Abnahme bei -70°C zu lagern.

3
3 Handhabung von Blutproben

4
4

5
5

6
6 Beispielmangel 1: Die Einhaltung der Vorgaben des Studienplans ist anhand der Rohdaten nicht rekonstruierbar. Die protokollkonforme Durchführung der Zentrifugation wurde nicht dokumentiert. Verwendetes Equipment wurde nicht dokumentiert. Zeitpunkt der Einlagerung in den TK wurde nicht dokumentiert.

7
7 Handhabung von Blutproben Was muss aufgezeichnet werden? Wer hat was, wann, wie und womit durchgeführt? Achtung: Mitunter keine Aufzeichnung in studienspezifischen Templates vorgesehen.

8
8 Handhabung von Blutproben Was muss aufgezeichnet werden… Ist rekonstruierbar, dass die Vorgaben des Protokolls bezüglich Probenaufarbeitung eingehalten wurden? Gilt es im Bezug auf die Handhabung der Proben besondere Lagerungsbedingungen einzuhalten (Temperatur, Licht)? Ist dokumentiert welche Geräte verwendet wurden (Zentrifugen, Messmittel)? Ist das verwendete Blutgerinnungsmittel dokumentiert?
. Ist dokumentiert welche Geräte verwendet wurden (Zentrifugen, Messmittel). Ist das verwendete Blutgerinnungsmittel dokumentiert .")
9
Mündliche Situationsbeschreibung Die Proben werden in einem -70°C Tiefkühler gelagert, welcher an eine zentrales Temperatur-überwachungssystem angeschlossen ist. Für den Zeitraum der Studie wurde keine Störung gemeldet. 9 Handhabung von Blutproben Beispielmangel 2: Die korrekte Lagerung der PK Proben ist nicht rekonstruierbar.

10
10 Handhabung von Blutproben Faktenbasierte Darstellung: Tiefkühler war als Leihgabe nicht vom Inventarisierungssystem und Wartungssystem des Krankenhauses erfasst. Der von der Haustechnik im Tiefkühler angebrachte Temperatursensor wurde im gesamten Studienzeitraum von 4 Jahren nie kalibriert. Keine ausreichende Leistungsqualifizierung des Überwachungssystems durchgeführt. Zum Zeitpunkt der Inspektion war der Tiefkühler defekt und außer Betrieb genommen, in der Leitstelle wurde uns gleichzeitig die korrekte Funktionsweise des Tiefkühlers bestätigt.

11
11 Handhabung von Blutproben Was ist die Stabilität einer Substanz? Substanzen unterliegen einem Abbau der in Abhängigkeit von der Zeit zu einer Konzentrations- änderungen führt. Einflussfaktoren Temperatur: Für niedermolekulare Verbindungen gilt: 10°C Temperaturerhöhung bedeutet doppelte Reaktionsgeschwindigkeit. Matrix: Der Abbau ist meist abhängig von der Matrix (Blut, Plasma, Lösungsmittel). Gefrier-Auftauzyklen
. Gefrier-Auftauzyklen.")
12
12 Handhabung von Blutproben Was muss aufgezeichnet werden? Ab wann wurde was, wo, wie und wie lange gelagert? Achtung: Mitunter keine Aufzeichnung in studienspezifischen Templates vorgesehen.

13
13 Handhabung von Blutproben Was muss aufgezeichnet werden: Wird der Einlagerungsort dokumentiert (Gerätekennung)? Wird der Einlagerungszeitpunkt (Uhrzeit und Tag) dokumentiert? Wartung und Reinigung durchgeführt und dokumentiert? Gibt es eine Temperaturaufzeichnung? Werden Temperaturaufzeichnungen archiviert? Wurden die Messmittel kalibriert? Gibt es ein Alarmsystem bei Temperaturüberschreitungen?
 dokumentiert. Wartung und Reinigung durchgeführt und dokumentiert. Gibt es eine Temperaturaufzeichnung. Werden Temperaturaufzeichnungen archiviert. Wurden die Messmittel kalibriert. Gibt es ein Alarmsystem bei Temperaturüberschreitungen .")
14
14 Handhabung von Blutproben Beispielmangel 3: Der Abnahmezeitpunkt der PK Proben wurde von Person A auf den Blutabnahmeröhrchen notiert. Der Abnahmezeitpunkt wurde von Person B in eine Liste übertragen und die Röhrchen unmittelbar danach verworfen, ohne Sicherstellung der Richtigkeit der übertragenen Werte.

15
15 Handhabung von Blutproben Wo ist die Aufzeichnung von Rohdaten vorgesehen? Rohdaten sind immer der erste Aufzeichnungsort ( elektronisch oder in Papier). Alles andere sind Überträge, Ausdrucke, Kopien.
. Alles andere sind Überträge, Ausdrucke, Kopien..")
16
16 Handhabung von Blutproben Was sonst noch zu beachten ist… Schulung von Studienpersonal Aufzeichnungen zu Wartung und Kalibrierung von Geräten Kennzeichnung von Blutproben Abpackung für Probenversand Dokumentation aller versandten Proben Maßnahmen zur Sicherstellung der Temperaturbedingungen beim Probenversand Dokumentation der Messmitteln für den Probentransport …

17
17 Handhabung von Blutproben Einhaltung der Vorgaben des Protokolls muss rekonstruierbar sein.

18
18 Handhabung von Prüfmedikation

19
19 Handhabung von Prüfmedikation

20
20 Was muss nachvollziehbar sein? Delegation der am Prüfzentrum verantwortlichen Personen Versand übernehmende Person Zeitpunkt der Übernahme Zuordenbare Temperaturaufzeichnungen Kontrolle der Versandbedingungen – ev. Quarantäne? Übernahmebestätigung an den Sponsor Handhabung von Prüfmedikation

21
21 Lagerung Geeigneter Lagerort Getrennte Lagerung von retournierter und unverbrauchter Medikation Temperaturkontolle - Verlauf (Min/Max Werte), in vorgesehenen Intervallen, vor Ausgabe! Bezug zu Lagerort, Temperaturmessgerät, Person Mittels qualifizierter Messgeräte – zentrale Temperaturüberwachung? Prozess bei Temperaturabweichungen Handhabung von Prüfmedikation

22
22 Accountability – laufend! Führung von Inventaraufzeichungen: Auskunft über vorliegende Prüfmedikationsmengen am Zentrum zu jedem Zeitpunkt der Studie Accountability auf PatientInnen/ProbandInnenebende: Was wurde wann von wem an welche PatientIn/ProbandIn ausgegeben(vorbereitet)/eingenommen (verabreicht)/retourniert. Übereinstimmung mit am Prüfzentrum vorhandener/retournierter Medikation Retournierung oder Vernichtung? - Prozess Handhabung von Prüfmedikation
/eingenommen (verabreicht)/retourniert. Übereinstimmung mit am Prüfzentrum vorhandener/retournierter Medikation Retournierung oder Vernichtung. - Prozess Handhabung von Prüfmedikation.")
23
23 Beispielmangel Versand : Nach Ankunft der Prüfmedikation am Prüfzentrum wurde der Logger nicht ausgelesen, sondern mit der Medikation in den Kühlschrank gelegt. 2 Tage später fiel dies dem Studienteam auf, eine Note to file wurde diesbezüglich verfasst, welche den tatsächlichen Sachverhalt nicht korrekt beschreibt (inkorrektes Datum,..). Bei Empfang der Prüfmedikation erfolgte keine zeitgerechte Kontrolle der Versandbedingungen. Der tatsächliche Sachverhalt ist über die am Prüfzentrum vorliegende Dokumentation nicht eindeutig nachvollziehbar. Es erfolgte vom zuständigen Monitor keine Information an den Sponsor. Handhabung von Prüfmedikation
. Bei Empfang der Prüfmedikation erfolgte keine zeitgerechte Kontrolle der Versandbedingungen. Der tatsächliche Sachverhalt ist über die am Prüfzentrum vorliegende Dokumentation nicht eindeutig nachvollziehbar. Es erfolgte vom zuständigen Monitor keine Information an den Sponsor. Handhabung von Prüfmedikation.")
24
24 Beispielmangel Lagerung: Die Temperaturkontrolle erfolgte entgegen der Sponsorvorgaben nur monatlich. Das verwendete Temperaturmessgerät ist auf den Temperaturaufzeichnungen nicht ersichtlich, Aufzeichnung der Aktualtemperatur. Die letzte Wartung des verwendeten Kühlschrankes ist nicht nachvollziehbar. Es liegt nur eine monatliche Dokumentation der Temperaturkontrolle vor, dies wurde vom zuständigen Monitor nicht beanstandet Eine Kontrolle der Aktualtemperatur ist nicht ausreichend (nicht nachvollziehbar, ob es zwischen den Auslesungen Abweichungen gab) - kalibriertes Min – Max –Thermometer! Die Temperaturaufzeichnungen zeigen keine Referenz zum verwendeten Messgerät Handhabung von Prüfmedikation
 - kalibriertes Min – Max –Thermometer. Die Temperaturaufzeichnungen zeigen keine Referenz zum verwendeten Messgerät Handhabung von Prüfmedikation.")
25
25 Beispielmangel Accountability - Inventaraufzeichnungen: Vereinzelt erfolgte die Eintragungen nicht in der zeitlichen Reihenfolge des Eintreffens der Lieferungen am Prüfzentrum Bestätigung des Eintreffens der Lieferung mit einem Datum vor dem tatsächlichen Versand Inkorrekte Angaben – Kits als ausgegeben vermerkt obwohl expired und nicht ausgegeben (am Prüfzentrum vorhanden oder retourniert) Kits doppelt eingetragen mit unterschiedlichem Ankunftsdatum Datum der Rücknahme an den Sponsor auf den Inventaraufzeichnungen entspricht nicht dem Datum der Retournierung auf den Versandunterlagen zur Retournierung Handhabung von Prüfmedikation
 Kits doppelt eingetragen mit unterschiedlichem Ankunftsdatum Datum der Rücknahme an den Sponsor auf den Inventaraufzeichnungen entspricht nicht dem Datum der Retournierung auf den Versandunterlagen zur Retournierung Handhabung von Prüfmedikation")
26
26 Danke für Ihre Aufmerksamkeit! clinicaltrials@ages.atclinicaltrials@ages.at Fragen

27
Austrian Agency for Health and Food Safetywww.ages.at Transparenzvorgaben in der Klinischen Prüfung Dr. Stefan Strasser Institut Überwachung, Abteilung Klinische Prüfung stefan.strasser@ages.at AGES Gespräch 29.10.2015

28
EudraCT Database Nationale Behörde Antragsteller PublicEMA/NCAs XML Local Drive Submission Package EudraCT DB EudraCT DWH EudraCT CTR EudraCT Public XML + Review 28 Stefan Strasser 29.10.2015

29
BASG Decisions 29 http://www.basg.gv.at/ueber-uns/basg-abstimmungen-und-verlautbarungen/http://www.basg.gv.at/ueber-uns/basg-abstimmungen-und-verlautbarungen/ BASG Abstimmungen BASG GZ Stefan Strasser 29.10.2015

30
EU Clinical Trials Register 30 https://www.clinicaltrialsregister.eu Stefan Strasser 29.10.2015

31
Publication of results “As of 21 July 2014, it will become mandatory for sponsors to post clinical trial results in the European Clinical trials Database (EudraCT), managed by the European Medicines Agency (EMA).” “Since the result-related information is fed into the publicly accessible European Union Clinical Trials Register, summary results of clinical trials will become available to the public as sponsors start to comply with their legal obligations.” https://eudract.ema.europa.eu/document.htmlhttps://eudract.ema.europa.eu/document.html „Result related documentation“ & „Training on EudraCT results“ 31 Stefan Strasser 29.10.2015
, managed by the European Medicines Agency (EMA). Since the result-related information is fed into the publicly accessible European Union Clinical Trials Register, summary results of clinical trials will become available to the public as sponsors start to comply with their legal obligations. „Result related documentation & „Training on EudraCT results 31 Stefan Strasser")
32
Legal Basis Article 11 of Directive 2001/20/EC Article 57(B) & 80 of Regulation EC/726/2004 Article 41 of Regulation EC/1901/2006 32 trials regulated by Directive 2001/20/EC (“Clinical Trials Directive”) trials regulated by Regulation EC/1901/2006 (“Paediatric Regulation”) Scope Stefan Strasser 29.10.2015
 & 80 of Regulation EC/726/2004 Article 41 of Regulation EC/1901/ trials regulated by Directive 2001/20/EC ( Clinical Trials Directive ) trials regulated by Regulation EC/1901/2006 ( Paediatric Regulation ) Scope Stefan Strasser")
33
EC/1901/2006 trials part of a paediatric investigation plan (PIP) (including non-paediatric trials and trials outside the EEA) or falling within Article 45 of EC/1901/2006 “ any paediatric studies already completed, by the date of entry into force, in respect of products authorised in the Community” or falling within Article 46 EC/1901/2006 “any other marketing authorisation holder-sponsored studies which involve the use in the paediatric population of a medicinal product covered by a marketing authorisation, whether or not they are conducted in compliance with an agreed paediatric investigation plan” Stefan Strasser 29.10.2015 33
 (including non-paediatric trials and trials outside the EEA) or falling within Article 45 of EC/1901/2006 any paediatric studies already completed, by the date of entry into force, in respect of products authorised in the Community or falling within Article 46 EC/1901/2006 any other marketing authorisation holder-sponsored studies which involve the use in the paediatric population of a medicinal product covered by a marketing authorisation, whether or not they are conducted in compliance with an agreed paediatric investigation plan Stefan Strasser")
34
Changes for NCA/IEC replaces submission to the NCA (4.3 CT-1) replaces submission to the IEC (4.2.1 CT-1) (for published information) Publication criteria: Result-related information on non-paediatric Phase-I clinical trials is not made public! Only non-paediatric Phase I trials still have to report to the IEC. 34 Stefan Strasser 29.10.2015

35
Full-Data-Set Contents Trial information - Study identification - Identifiers - Sponsor details - Paediatric regulatory details - Result analysis stage - General Information about the trial - Population of trial subjects with actual number of subjects included in the trial Trial information - Study identification - Identifiers - Sponsor details - Paediatric regulatory details - Result analysis stage - General Information about the trial - Population of trial subjects with actual number of subjects included in the trial Subject disposition - Recruitment - Pre-assignment Period - Post-Assignment Periods Subject disposition - Recruitment - Pre-assignment Period - Post-Assignment Periods Baseline Characteristics - Age - Gender - Study Specific Baseline Characteristics - Age - Gender - Study Specific End Points - Endpoint definitions -- End Point #1 --- Statistical Analyses -- End Point #2, --- Statistical Analyses … End Points - Endpoint definitions -- End Point #1 --- Statistical Analyses -- End Point #2, --- Statistical Analyses … Adverse Events - Adverse events information - Adverse event reporting group - Serious Adverse Events - Non-serious adverse event Adverse Events - Adverse events information - Adverse event reporting group - Serious Adverse Events - Non-serious adverse event More Information - Global Substantial Amendments - Global Interruptions and re-starts - Limitations & Caveats More Information - Global Substantial Amendments - Global Interruptions and re-starts - Limitations & Caveats Stefan Strasser 29.10.2015 35

36
Under construction… Stefan Strasser 29.10.2015 36

37
2001/20/EC adult trials End of trialComposition of resultsTiming of posting On/After FoP Full data set mandatory, summary attachment(s) optional 12 months after EOT < 1 year prior to FoP Full data set mandatory, summary attachment(s) optional 12 months after FoP ≥ 1 year prior to FoP Full data set or summary or both 24 months after FoP All non-paediatric trials conducted in at least one EEA country, whether or not included in an agreed paediatric investigation plan (PIP). Finalisation of programming (FoP) = 21 July 2014 Stefan Strasser 29.10.2015 37
 = 21 July 2014 Stefan Strasser")
38
2001/20/EC paediatric trials End of trialComposition of resultsTiming of posting On/After FoP Full data set mandatory, summary attachment(s) optional 6 months after EOT 12 months (if justified) < 1 year prior to FoP Full data set mandatory, summary attachment(s) optional 12 months after FoP ≥ 1 year prior to FoP Full data set or summary or both 24 months after FoP Paediatric trials completed after 26 January 2007, conducted in at least one EEA country and not being marketing authorisation holder-sponsored. Finalisation of programming (FoP) = 21 July 2014 Stefan Strasser 29.10.2015 38
 = 21 July 2014 Stefan Strasser")
39
Mitigating Measures „Summary attachment“: - a copy, authorised by the copy right-holder, of a medical journal article (as PDF file), - the synopsis in accordance with Annex I to the ICH E3 guidance (as PDF file), -or any other appropriate document containing the information of that synopsis (as PDF file) „Justification of 12 months“ -the clinical trial does not fall within the scope of Article 46 of the Paediatric Regulation, and -it is for objective scientific reasons not possible to submit the result-related information within six months, which has been demonstrated by the submitting party Stefan Strasser 29.10.2015 39
, - the synopsis in accordance with Annex I to the ICH E3 guidance (as PDF file), -or any other appropriate document containing the information of that synopsis (as PDF file) „Justification of 12 months -the clinical trial does not fall within the scope of Article 46 of the Paediatric Regulation, and -it is for objective scientific reasons not possible to submit the result-related information within six months, which has been demonstrated by the submitting party Stefan Strasser")
40
ICH E3 Annex I Stefan Strasser 29.10.2015 40

41
EC/1901/2006 trials Art. 46 End of trialComposition of resultsTiming of posting On/After FoP Full data set mandatory, summary attachment(s) optional 6 months after EOT Prior to FoP Full data set mandatory, summary attachment(s) optional 12 months after FoP Paediatric trials completed after 26 January 2007 which involve the use of a medicinal product covered by an EU marketing authorisation and sponsored by the marketing authorisation holder, whether or not included in an agreed PIP. Finalisation of programming (FoP) = 21 July 2014 Stefan Strasser 29.10.2015 41
 optional 6 months after EOT Prior to FoP Full data set mandatory, summary attachment(s) optional 12 months after FoP Paediatric trials completed after 26 January 2007 which involve the use of a medicinal product covered by an EU marketing authorisation and sponsored by the marketing authorisation holder, whether or not included in an agreed PIP. Finalisation of programming (FoP) = 21 July 2014 Stefan Strasser")
42
EC/1901/2006 trials Art. 45 End of trialComposition of resultsTiming of posting Prior to 26.01.2007 Summary of study submitted to EMA and uploaded by the Agency for publication 24 months after FoP Paediatric trials in respect of products covered by an EU marketing authorisation on 26 January 2007, completed by 26 January 2007.. Finalisation of programming (FoP) = 21 July 2014 Stefan Strasser 29.10.2015 42
 = 21 July 2014 Stefan Strasser")
43
EC/1901/2006 trials Art. 41(1) End of trialComposition of resultsTiming of posting On/After FoP Full data set mandatory, summary attachment(s) optional 6 months after EOT 12 months if justified Prior to FoP Full data set mandatory, summary attachment(s) optional 12 months after FoP Paediatric trials included in an agreed PIP, not sponsored by the marketing authorisation holder and conducted fully outside the EEA. Finalisation of programming (FoP) = 21 July 2014 Stefan Strasser 29.10.2015 43
 End of trialComposition of resultsTiming of posting On/After FoP Full data set mandatory, summary attachment(s) optional 6 months after EOT 12 months if justified Prior to FoP Full data set mandatory, summary attachment(s) optional 12 months after FoP Paediatric trials included in an agreed PIP, not sponsored by the marketing authorisation holder and conducted fully outside the EEA. Finalisation of programming (FoP) = 21 July 2014 Stefan Strasser")
44
Reporting modalities If the clinical trial ends prematurely, that date should be considered the end of the trial. Only one set of result-related data may be provided per planned analysis and trial. If the outcome is analysed on several occasions, each of these analyses should be posted. Information should be posted in English. In addition, the information may be posted in any other official EU language Stefan Strasser 29.10.2015 44

45
Publication The posted result-related information is made public through the EU Clinical Trials Register (only result-related information on non- paediatric Phase-I clinical trials is not made public.) The result-related information is made public within 15 working days from the posting of a valid data set. In addition to being readable in situ on the web, the data will also be made available in a printable format and in a downloadable format. The web interface is going to provide tools to facilitate the searching, reading and browsing of the public information on clinical trials and their results. Stefan Strasser 29.10.2015 45

46
Non-compliance & factual inaccuracy Member States should verify that for clinical trials authorised by them the result-related information is posted to the Agency. Clinical trials for which no result-related information has been posted 9 months after the end of the trial for paediatric trials or 15 months for other trials will be flagged. This information will be publicly available. The anticipated duration of the trial is entered at the time of the clinical trial application. All corrections to published information will be made by the party posting that information, sometimes upon request by the Agency. Stefan Strasser 29.10.2015 46

47
Transparency in EC 536/2014 Characteristics of all clinical trials (Phase I-IV) Summary report, summary for laypersons and clinical study report (in case of marketing authorisation application) Protocol and Patient Information Sheet All study documents except quality aspects Information on trial sites and investigators Inspections and inspection reprots Corrective actions by the authorities … Stefan Strasser 29.10.2015 47
 Summary report, summary for laypersons and clinical study report (in case of marketing authorisation application) Protocol and Patient Information Sheet All study documents except quality aspects Information on trial sites and investigators Inspections and inspection reprots Corrective actions by the authorities … Stefan Strasser")
48
Thank you for your attention! Questions? Stefan Strasser 29.10.2015 48

49
Austrian Agency for Health and Food Safetywww.ages.at Einstufung von AMG Studien Dr. Stefan Strasser Institut Überwachung, Abteilung Klinische Prüfung stefan.strasser@ages.at AGES Gespräch 29.10.2015

50
Warum? Häufige Anfragen über clinicaltrials@ages.at und nis@ages.at clinicaltrials@ages.atnis@ages.at Zahlreiche Konsequenzen für den Antragsteller Ethikkommissionen verweisen Antragsteller an das BASG Stefan Strasser, 29.10.2015 50

51
51 Wo zu finden? Scroll Down http://www.basg.gv.at/arzneimittel/vor-der-zulassung/klinische-pruefungen/ Stefan Strasser, 29.10.2015

52
Rechtliche Grundlagen § 2a. (1) AMG „Klinische Prüfung“ (KP) § 2a. (3) AMG „Nicht-interventionelle Studie“ (NIS) EC Question & Answers v10.0 (April 2012) 52 www.office.com Stefan Strasser, 29.10.2015
 AMG „Nicht-interventionelle Studie (NIS) EC Question & Answers v10.0 (April 2012) 52 Stefan Strasser,")
53
Frage 1 Studie am Menschen? AMG: „Untersuchung an einem Prüfungsteilnehmer“ Anlehnung an die Deklaration von Helsinki Ausnahmen: -Studien an Tieren, Pflanzen, Mineralien… -Studien an Leichen 53 www.office.com Stefan Strasser, 29.10.2015

54
Frage 2 „Prospektiv“ oder „Retrospektiv“? Begriffe nicht im AMG definiert Auslegung im Sinne des „Schutzaspektes“ neue Daten? “Retrospektiv”: -alle notwendigen Daten/Proben bereits vorhanden -„Aktenschrank“ oder „Kühlschrank“ 54 www.office.com Stefan Strasser, 29.10.2015

55
Frage 3 Substanzeinnahme? Substanz AMG §1. (4) „Stoff” Auch vor Beginn der Studie! Auch „Routineanwendungen“! Ausnahme: Keine Selektion oder Auswertung gemäß Substanzeinnahme 55 www.office.com Stefan Strasser, 29.10.2015

56
Frage 4 Arzneimittel? §1. (1) und (2) und (3) AMG „Funktionsarzneimittel“ „Präsentationsarzneimittel“ Eine Befassung des Abgrenzungsbeirates wird empfohlen! 56 www.office.com Stefan Strasser, 29.10.2015
 und (2) und (3) AMG „Funktionsarzneimittel „Präsentationsarzneimittel Eine Befassung des Abgrenzungsbeirates wird empfohlen Stefan Strasser,")
57
Frage 5 Korrelation Einnahme - Endpunkt? „Wirkung“ umfasst „Wirksamkeit“ („Efficacy“) und „Pharmakodynamik“ („Effects“) Gilt auch für Vergleichsarme (z.B. in MPG Studien) Ausnahmen: ausschließlich andere Endpunkte (z.B. Verschreibungsverhalten) 57 www.office.com Stefan Strasser, 29.10.2015
 und „Pharmakodynamik („Effects ) Gilt auch für Vergleichsarme (z.B. in MPG Studien) Ausnahmen: ausschließlich andere Endpunkte (z.B. Verschreibungsverhalten) 57 Stefan Strasser,")
58
Klinische Prüfung oder NIS? Alle Kriterien des §2a. (3) müssen erfüllt sein NIS Leitfaden auf der BASG Website Häufige Probleme: Arzneispezialität muss in Österreich zugelassen sein Alle Maßnahmen müssen für die individuelle klinische Situation Standard und nicht zusätzlich belastend sein Randomisierung oder Zuteilung in Gruppen Begriffe wie „Beobachtungsstudien“, „Register“, „PASS“* *Post Authorization Safety Studies 58 Stefan Strasser, 29.10.2015
 müssen erfüllt sein NIS Leitfaden auf der BASG Website Häufige Probleme: Arzneispezialität muss in Österreich zugelassen sein Alle Maßnahmen müssen für die individuelle klinische Situation Standard und nicht zusätzlich belastend sein Randomisierung oder Zuteilung in Gruppen Begriffe wie „Beobachtungsstudien , „Register , „PASS * *Post Authorization Safety Studies 58 Stefan Strasser,")
59
Austrian Agency for Health and Food Safetywww.ages.at Fallbeispiele 59

60
Exkurs „Biomarker“ 60 Biomarker allgemein -charakteristisches biologisches Merkmal -objektiv messbar -deutet auf normalen oder krankhaften Prozess im Körper oder die Reaktion auf eine therapeutische Intervention hin -Zellen, Gene, Genprodukte oder bestimmte Moleküle wie Enzyme oder Hormone -komplexe Organfunktionen oder charakteristische Veränderungen biologischer Strukturen Krankheitsbezogene Biomarker -diagnostisch (ob eine Erkrankung besteht) -prognostisch (ob und wie sich eine Krankheit entwickelt) Arzneimittelbezogene Biomarker geben Hinweis über Wirksamkeit, Sicherheit oder Kinetik bei einem Patienten/ einer Patientengruppe www.office.com Stefan Strasser, 29.10.2015
 -prognostisch (ob und wie sich eine Krankheit entwickelt) Arzneimittelbezogene Biomarker geben Hinweis über Wirksamkeit, Sicherheit oder Kinetik bei einem Patienten/ einer Patientengruppe Stefan Strasser,")
61
www.office.com Biomarker 1 AM* Untersuchung in-vitro (Tumormodell) Tumorproben vom Patienten Prospektive Probenentnahme bei der Operation Arzneimittel wird nur in-vitro zugesetzt Wirkung auf Tumorzellen wird untersucht NIS n.a. Keine Studie nach AMG *AM = Arzneimittel 61 Stefan Strasser, 29.10.2015

62
www.office.com Biomarker 2 Biomarker in Restproben aus klinischer Routine Tumorproben vom Patienten Probenentnahme bereits im Rahmen der Routine (?) gesammelt (retrospektiv) Arzneimitteleinnahme im Rahmen der Routine Korrelation Tumormarker Therapieerfolg NIS n.a. Keine Studie nach AMG 62 Stefan Strasser, 29.10.2015

63
www.office.com Biomarker 3 Therapiebezogener Biomarker bei zugelassener Therapie (z.B. Multiple Sklerose) Klinische Prüfung nach 2a. (1) AMG 63 Patienten Prospektive Datensammlung & Probenentnahme Arzneimitteleinnahme im Rahmen der Routine Korrelation Zielparameter klinischer Outcome Keine NIS, da zusätzliche Maßnahme Stefan Strasser, 29.10.2015
 Klinische Prüfung nach 2a. (1) AMG 63 Patienten Prospektive Datensammlung & Probenentnahme Arzneimitteleinnahme im Rahmen der Routine Korrelation Zielparameter klinischer Outcome Keine NIS, da zusätzliche Maßnahme Stefan Strasser,")
64
www.office.com Nahrungsergänzungsmittel 1 Studie mit Vitamin D Biogena® zur Vitamin-D- Supplementierung (≤TUI*) Studienteilnehmer mit Vitamin-D-Mangel Prospektive Datensammlung Einnahme von Vitamin D in Dosis ≤TUI Korrelation Einnahme klinischer Outcome NIS n.a. Keine Studie nach AMG *tolerable upper intake level (dzt. 4,000 IE/d) 64 Stefan Strasser, 29.10.2015
 64 Stefan Strasser,")
65
www.office.com Nahrungsergänzungsmittel 2 Studie von Vitamin-D-Biogena® zur Therapie (>TUI*) einer kardiovaskulären Erkrankung Klinische Prüfung nach 2a. (1) AMG? 65 Patienten mit Erkrankung Prospektive Datensammlung Einnahme von Vitamin D in Dosis >>> TUI Korrelation Einnahme klinischer Outcome Keine NIS, da kein zugelassenes AM *tolerable upper intake level (dzt. 4,000 IE/d) Stefan Strasser, 29.10.2015
 AMG. 65 Patienten mit Erkrankung Prospektive Datensammlung Einnahme von Vitamin D in Dosis >>> TUI Korrelation Einnahme klinischer Outcome Keine NIS, da kein zugelassenes AM *tolerable upper intake level (dzt. 4,000 IE/d) Stefan Strasser,")
66
www.office.com Diagnostika 1 Untersuchung von Anatomie oder Physiologie durch ein AM Patienten mit Tumorerkrankung Prospektive Datensammlung Anwendung eines PET Tracers (ohne Zulassung) Anatomie/Physiologie des Tumors NIS n.a. keine Studie nach AMG (da AM Mittel zum Zweck und nicht Gegenstand der Untersuchung) 66 Stefan Strasser, 29.10.2015
 66 Stefan Strasser,")
67
www.office.com Diagnostika 2 Untersuchung eines diagnostischen AM Patienten mit Tumorerkrankung Prospektive Datensammlung Anwendung eines PET Tracers (ohne Zulassung) diagnostische Eigenschaften des PET Tracers keine NIS, da keine Zulassung Klinische Prüfung nach 2a. (1) AMG (da AM Gegenstand der Untersuchung) 67 Stefan Strasser, 29.10.2015
 AMG (da AM Gegenstand der Untersuchung) 67 Stefan Strasser,")
68
www.office.com Arzneimittel als Komparatoren Randomisierte Studie Doppelballon-Einleitung vs. Dinoproston (Standardtherapie) Patientinnen Indikation zur Geburtseinleitung Prospektive Datensammlung Einnahme des AM im Vergleichsarm (Standard) Korrelation mit Wirksamkeit/Sicherheit Keine NIS wegen Randomisierung Klinische Prüfung nach 2a. (1) AMG ( MPG!) 68 Stefan Strasser, 29.10.2015
 Patientinnen Indikation zur Geburtseinleitung Prospektive Datensammlung Einnahme des AM im Vergleichsarm (Standard) Korrelation mit Wirksamkeit/Sicherheit Keine NIS wegen Randomisierung Klinische Prüfung nach 2a. (1) AMG ( MPG!) 68 Stefan Strasser,")
69
www.office.com Nicht-zulassungspflichtige AM Erfassung von Sicherheitsdaten nach speziellen Formen der Bluttransfusion Patienten mit Indikation zur Transfusion Prospektive Datensammlung Transfusion im Rahmen der Routine Systematische Untersuchung der Sicherheit Keine NIS, da keine Zulassungsfähigkeit für Blutprodukte Klinische Prüfung nach 2a. (1) AMG 69 Stefan Strasser, 29.10.2015
 AMG 69 Stefan Strasser,")
70
Austrian Agency for Health and Food Safetywww.ages.at Danke für die Aufmerksamkeit! Fragen?

71
Austrian Agency for Health and Food Safetywww.ages.at Dr. med. Violetta Zmuda AGES MEA, Institut Überwachung, Abteilung Klinische Prüfung AGES-Gespräch Wien, 29. Oktober 2015 LEITFADEN Klinische Prüfung von Medizinprodukten (MP) und Leistungsbewertungsprüfung von In-vitro-Diagnostika (IVD) „Einreichung und Durchführung“ NEU! www.basg.at
 und Leistungsbewertungsprüfung von In-vitro-Diagnostika (IVD) „Einreichung und Durchführung NEU.")
72
72 Wo wird der neue Leitfaden zu finden sein? – www.basg.gv.at

73
73 1. Voraussetzungen für eine KP eines MP / eine LBP eines IVDs 2. Meldeverfahren 3. Ordnungsgemäße Meldung 4. Bearbeitung seitens des BASG 5. Protokolländerungen (Amendments) 6. Meldepflichten während einer Klinischen Prüfung 7. Meldepflichten nach der Klinischen Prüfung 8. AMG/MPG-Studien (Kombistudien) 9. Gebühren Inhalt des Leitfadens www.basg.at
 6. Meldepflichten während einer Klinischen Prüfung 7. Meldepflichten nach der Klinischen Prüfung 8. AMG/MPG-Studien (Kombistudien) 9. Gebühren Inhalt des Leitfadens")
74
74 Definition der KP/ LBP gemäß MPG ist erfüllt, wenn Prüfprodukt ist ein MP bzw. ein IVD gemäß Definition des MPG und soll systematisch an Prüfungsteilnehmern untersucht werden ODER Prüfprodukt ist kein MP bzw. IVD, soll jedoch systematisch an Prüfungsteilnehmern untersucht werden und die Zielsetzung des Forschungsvorhabens erfüllt die Definition eines Medizinprodukts / IVD‘s gemäß MPG 1. Voraussetzungen für eine KP eines MP / eine LBP eines IVDs www.basg.at

75
75 Wenn einer der folgenden Punkte zutrifft: Prüfprodukt ohne CE-Kennzeichnung Prüfprodukt mit CE-Kennzeichnung, jedoch Anwendung außerhalb der Zweckbestimmung Anwendung innerhalb der Zweckbestimmung, jedoch zusätzliche diagnostische oder therapeutische Maßnahmen Aktives implantierbares Medizinprodukt (AIMD) Prototyp - Generierung klinischer Daten „Pilotstudien“, „proof-of-concept“-Studien, „Grundlagenforschung“: MP bzw. IVD Gegenstand der Untersuchung Ergebnisse Entwicklung eines MP Studie - Generierung von Studienergebnissen Modifikation der IFU 1.1. Wann ist die Meldung einer KP an die Behörde erforderlich? www.basg.at

76
76 Ein zutreffender Punkt ist ausreichend: In-vitro-Diagnostikum ohne CE-Kennzeichnung Zusätzl. studienbezogene Probenahme (Art, Volumen, Dauer) Zusätzl. medizinische Untersuchungen oder Behandlungen In-vitro-Diagnostikum soll entwickelt, optimiert bzw. validiert werden Studie - Ergebnisse generieren Modifikation der Gebrauchsanweisung Die Zuständigkeit der AT Behörde ergibt sich durch den Ort der Probennahme, nicht der Analyse österreichische Prüfungsteilnehmer bzw. Proben Zuständigkeit AT 1.2. Wann ist die Meldung einer LBP an die Behörde erforderlich? www.basg.at
 Zusätzl. medizinische Untersuchungen oder Behandlungen In-vitro-Diagnostikum soll entwickelt, optimiert bzw. validiert werden Studie - Ergebnisse generieren Modifikation der Gebrauchsanweisung Die Zuständigkeit der AT Behörde ergibt sich durch den Ort der Probennahme, nicht der Analyse österreichische Prüfungsteilnehmer bzw. Proben Zuständigkeit AT 1.2. Wann ist die Meldung einer LBP an die Behörde erforderlich.")
77
77 2. Meldeverfahren Das Medizinproduktegesetz legt für die Beurteilung von Klinischen Prüfungen durch das Bundesamt für Sicherheit im Gesundheitswesen folgende Verfahren fest: www.basg.at

78
78 Formular F_I202_Meldung einer klinischen Prüfung bzw. F_I282_Meldung einer Leistungsbewertungsprüfung Pro Prüfzentrum ausgefülltes Formular F_I204_Beiblatt Prüfzentrum bzw. F_I283_Beiblatt Leistungsbewertungsprüfung Unterlagen gemäß der Liste L_I07_Notwendige Unterlagen für eine Klinische Prüfung bzw. L_I63_Liste „Notwendige Unterlagen für eine Leistungsbewertungsprüfung Elektronisches Meldeformular in Entwicklung!! 3. Ordnungsgemäße Meldung Zu finden unter: http://www.basg.gv.at/medizinprodukte/klinische-pruefung-von-medizinprodukten www.basg.at

79
79 Einfache Ausfertigung in Papierform und in elektronischer Form (CD-Rom/Email) vor Beginn der Klinischen Prüfung Adresse Bundesamt für Sicherheit im Gesundheitswesen (BASG) Österreichische Agentur für Gesundheit und Ernährungssicherheit(AGES) Institut Überwachung (INS), Abteilung Klinische Prüfung (CLTR) Traisengasse 5, A-1200 Wien Meldeformulare sind signiert vorzulegen Eingangsbestätigung – ACHTUNG! automatisierte Bestätigung der Email Eingangsbestätigung der Unterlagen durch die Administratoren 3.1. In welcher Form muss die Meldung eingebracht werden? www.basg.at

80
80 Inhaltliche / fachliche Beurteilung technische Sicherheit die Plausibilität der eingereichten Unterlagen die medizinischen und wissenschaftlichen Daten und Informationen die Risiken in Abwägung zum voraussichtlichen Nutzen der klinischen Prüfung (Risikoanalyse) Mängelschreiben gemäß § 13 Abs. 3 AVG Arten der Bewilligung Nicht-Untersagung (§ 40 Abs. 2) Bescheid (§ 40 Abs. 2) Bestätigung ordnungsgemäßer Meldung (§ 40 Abs. 3, § 65a Abs. 2 MPG) evtl. Untersagung (gemäß § 41 Abs. 4 MPG) 4. Bearbeitung seitens des BASG www.basg.at
 Bescheid (§ 40 Abs. 2) Bestätigung ordnungsgemäßer Meldung (§ 40 Abs. 3, § 65a Abs. 2 MPG) evtl. Untersagung (gemäß § 41 Abs. 4 MPG) 4. Bearbeitung seitens des BASG")
81
81 Nur substantielle Protokoll-Änderungen sind vom Sponsor an das BASG zu melden! Auswirkung auf die Sicherheit oder die physische und mentale Unversehrtheit der Prüfungsteilnehmer ODER Beeinflussung der wissenschaftlichen Aussagekraft der klinischen Prüfung Deklaration obliegt dem Sponsor Einstufung: Liste „Klassifizierung von Amendments“ (Annex III) Amendment-Meldeformular, Übersichtliche Zusammenfassung der Änderungen, Begründung und geänderte Studienunterlagen in Papierform oder elektronisch an clinicaltrials@ages.at clinicaltrials@ages.at Vorgenommene Änderungen: klar erkenntlich machen! track-changes, farbliche Markierung, o.ä. 5. Protokolländerungen (Amendments) www.basg.at
 Amendment-Meldeformular, Übersichtliche Zusammenfassung der Änderungen, Begründung und geänderte Studienunterlagen in Papierform oder elektronisch an Vorgenommene Änderungen: klar erkenntlich machen. track-changes, farbliche Markierung, o.ä. 5. Protokolländerungen (Amendments)")
82
82 Meldepflichten des Sponsors § 42 Abs. 8 MPG Meldung von schwerwiegenden unerwünschten Ereignissen (Serious Adverse Events, SAEs) Meldepflichten des Klinischen Prüfers § 61 MPG Meldung aller schwerwiegender Nebenwirkungen an die Ethikkommission (unverzüglich) Die Meldepflichten des § 70 MPG bleiben unberührt § 64 Abs. 5 MPG Meldung aller Medizinproduktenebenwirkungen und aller schwerwiegenden unerwünschten Ereignisse an den Sponsor 6. Meldepflichten während einer Klinischen Prüfung www.basg.at
 Meldepflichten des Klinischen Prüfers § 61 MPG Meldung aller schwerwiegender Nebenwirkungen an die Ethikkommission (unverzüglich) Die Meldepflichten des § 70 MPG bleiben unberührt § 64 Abs. 5 MPG Meldung aller Medizinproduktenebenwirkungen und aller schwerwiegenden unerwünschten Ereignisse an den Sponsor 6. Meldepflichten während einer Klinischen Prüfung")
83
83 Verwendung der Formulare F_I208 bzw. F_I287: www.basg.at/medizinprodukte/formulare/klinische-pruefung/ F_I208 SAE Klinische Prüfung MP Meldeformular für jedes schwerwiegende unerwünschte Ereignis an einem Prüfzentrum in Österreich F_I287 SAE EWR Meldung MP Studie Fortlaufendes EU-Meldeformular (tabellarische Auflistung, Line Listing) Meldefristen (unverzüglich!) MEDDEV Guideline 2.7/3 „Clinical Investigation: serious adverse event reporting“ http://ec.europa.eu/DocsRoom/documents/10330/attachments/1/translations/en/renditions/native Innerhalb von zwei Kalendertagen: Schwerwiegende unerwünschte Ereignisse, welche ein unmittelbares Sterberisiko, eine schwerere Verletzung oder Krankheit verursachen Binnen sieben Kalendertagen: alle übrigen Ereignisse SAE-Meldung gemäß § 42 Abs. 8 MPG www.basg.at
 Meldefristen (unverzüglich!) MEDDEV Guideline 2.7/3 „Clinical Investigation: serious adverse event reporting Innerhalb von zwei Kalendertagen: Schwerwiegende unerwünschte Ereignisse, welche ein unmittelbares Sterberisiko, eine schwerere Verletzung oder Krankheit verursachen Binnen sieben Kalendertagen: alle übrigen Ereignisse SAE-Meldung gemäß § 42 Abs. 8 MPG")
84
84 Beendigung: unter Verwendung des dafür vorgesehenen Formulars F_I206 www.basg.at/medizinprodukte/formulare/klinische-pruefung/ Vorzeitige Beendigung: § 40 Abs. 6 MPG – Meldung vom Sponsor an das BASG und die zuständigen Behörden anderer betroffener Vertragsparteien des EWR (inkl. Begründung) Abschlussbericht: vom Sponsor zu erstellen und von allen an der Prüfung beteiligten Klinischen Prüfern zu unterzeichnen Kritische Bewertung der gewonnenen wissenschaftlich relevanten Daten (§ 46 Abs. 2 MPG) 7. Meldepflichten nach der Klinischen Prüfung www.basg.at
 Abschlussbericht: vom Sponsor zu erstellen und von allen an der Prüfung beteiligten Klinischen Prüfern zu unterzeichnen Kritische Bewertung der gewonnenen wissenschaftlich relevanten Daten (§ 46 Abs. 2 MPG) 7. Meldepflichten nach der Klinischen Prüfung")
85
85 Vorgaben für die Klinische Prüfung nach MPG und AMG beachten Im jeweiligen Anschreiben (Coverletter) als Kombistudie erkennbar machen Papiereinreichung – 2 Exemplare des Antrags Einmalige Einreichung aller elektronischen Unterlagen auf Datenträger Für Meldungen von Änderungen am Prüfplan (substantielle Amendments) und Meldungen von unerwünschten Ereignissen/Nebenwirkungen jeweilige Bestimmungen des MPG bzw. AMG einhalten getrennte Meldungen vornehmen Siehe Leitfaden zu AMG Studien: www.basg.at/arzneimittel/vor-der-zulassung/klinische-pruefungen/ www.basg.at/arzneimittel/vor-der-zulassung/klinische-pruefungen/ 8. AMG/MPG-Studien (Kombistudien) www.basg.at
")
86
86 Verordnung des BASG über den Gebührentarif gemäß Gesundheits- und Ernährungssicherheitsgesetzes: Gebühren: 2500 € Klinische Prüfung / Leistungsbewertungsprüfung 400 € Bedeutsame Änderung (substantielles Amendment) Bei zeitgleichem Einbringen einer Kombinationsstudie: MPG: volle Gebühr, sowie AMG: 35% der Gebühr Ausnahmen: akademische Klinische Studien Zurückziehung des Antrags -vor der Bestätigung der ordnungsgemäßen Meldung: 10% der Gebühr -zu einem späteren Zeitpunkt (inhaltliche Prüfung begonnen): gesamte Gebühr 9. Gebühren www.basg.at

87
87 Nur bestimmte Klinische Prüfungen/Leistungsbewertungsprüfungen sind an das BASG zu melden Die Ziele des Projektes und der Status der zur Anwendung kommenden Medizinprodukte definieren, ob die Studie dem MPG unterliegt Der Sponsor trägt die Verantwortung für die Einstufung, ob ein Medizinprodukt vorliegt die Einstufung des Projektes Ein kostenpflichtiger Antrag auf Abgrenzung des Produktes durch das BASG ist möglich medizinprodukte@ages.at Take-Home Messages… www.basg.at

88
Austrian Agency for Health and Food Safetywww.ages.at VIELEN DANK für die Aufmerksamkeit! www.basg.at

89
89 Für weitere Fragen zu MPG Aspekten clinical.trials@ages.atclinical.trials@ages.at, violetta.zmuda@ages.at, svetlana.seiter@ages.atvioletta.zmuda@ages.atsvetlana.seiter@ages.at Mehr Informationen http://www.basg.gv.at/medizinprodukte/klinische-pruefung-von-medizinprodukten http://www.basg.gv.at/medizinprodukte/klinische-pruefung-von-medizinprodukten www.basg.at

90
Austrian Agency for Health and Food Safetywww.ages.at Dr. Corina Spreitzer Institut Überwachung, Abteilung Klinische Prüfungen AGES-Gespräch Wien, 29.10.2015 Wissenswertes zu den jährlichen Sicherheitsberichten bzw. DSURs

91
91 Der Inhalt des Vortrags gibt die Sichtweisen der Vortragenden wieder und repräsentiert nicht offizielle Ansichten der AGES Medizinmarktaufsicht (MEA). Disclaimer

92
92 Directive 2001/20/EC Article 17 u. 18 AMG §41e.(3): Einmal jährlich während der gesamten Dauer der klinischen Prüfung hat der SPONSOR eine Liste mit allen mutmaßlichen Nebenwirkungen, die während der gesamten Prüfungsdauer aufgetreten sind, sowie einen Bericht über die Sicherheit der Prüfungsteilnehmer vorzulegen. Wem? Dem BASG und den zuständigen Behörden aller Vertragsparteien des EWR, in deren Hoheitsgebiet die klinische Prüfung durchgeführt wird, und den betreffenden Ethikkommissionen = Annual Safety Report (ASR) bzw. seit September 2011 gemäß ICH Guideline E2F Development Safety Update Report (DSUR) Rechtliche Grundlagen
: Einmal jährlich während der gesamten Dauer der klinischen Prüfung hat der SPONSOR eine Liste mit allen mutmaßlichen Nebenwirkungen, die während der gesamten Prüfungsdauer aufgetreten sind, sowie einen Bericht über die Sicherheit der Prüfungsteilnehmer vorzulegen. Wem. Dem BASG und den zuständigen Behörden aller Vertragsparteien des EWR, in deren Hoheitsgebiet die klinische Prüfung durchgeführt wird, und den betreffenden Ethikkommissionen = Annual Safety Report (ASR) bzw. seit September 2011 gemäß ICH Guideline E2F Development Safety Update Report (DSUR) Rechtliche Grundlagen.")
93
93 http://www.ich.org/products/guidelines/efficacy/article/ efficacy-guidelines.html

94
94 Der Sponsor ist verantwortlich für: - Erstellung - Inhalt - Einreichung Erstellung des DSUR kann delegiert werden Einreichung bei der/den zuständigen Behörden innerhalb von 60 Tagen nach Data Lock Point (DLP) ICH E2F – Verantwortung des Sponsors
 ICH E2F – Verantwortung des Sponsors")
95
95 All sections should be completed; when no information is available, this should be stated. Title page Executive Summary 1. Introduction 2. Worldwide Marketing Approval Status 3. Actions Taken in the Reporting Period for Safety Reasons 4. Changes to Reference Safety Information 5. Inventory of Clinical Trials Ongoing and Completed during the Reporting Period 6. Estimated Cumulative Exposure 6.1 Cumulative Subject Exposure in the Development Program 6.2 Patient Exposure from Marketing Experience 7. Data in Line Listings and Summary Tabulations 7.1 Reference Information 7.2 Line Listings of Serious Adverse Reactions during the Reporting Period 7.3 Cumulative Summary Tabulations of Serious Adverse Events DSUR example for non-commercial sponsors - TABLE OF CONTENTS I

96
96 8. Significant Findings from Clinical Trials during the Reporting Period 8.1 Completed Clinical Trials 8.2 Ongoing Clinical Trials 8.3 Long-term Follow-up 8.4 Other Therapeutic Use 8.5 New Safety Data Related to Combination Therapies 9. Safety Findings from Non-interventional Studies 10. Other Clinical Trial/Study Safety Information 11. Safety Findings from Marketing Experience 12. Non-clinical data 13. Literature 14. Other DSURs 15. Lack of Efficacy 16. Region-Specific Information 17. Late-Breaking Information 18. Overall Safety Assessment 18.1 Evaluation of the Risks 18.2 Benefit-risk considerations 19. Summary of Important risks 20. Conclusions Appendices DSUR example for non-commercial sponsors - TABLE OF CONTENTS II

97
97 Description of significant actions related to safety by: - the sponsor, - regulators, - data monitoring committees (DMC) - ethics committees The reason(s) for each action should be provided (if known). Relevant updates to previous actions (e.g., resumption of a clinical trial after suspension) Changes to IB should be discussed in section 3.4 “Changes to Reference Safety Information” Actions Taken in the Reporting Period for Safety Reasons
 Changes to IB should be discussed in section 3.4 Changes to Reference Safety Information Actions Taken in the Reporting Period for Safety Reasons.")
98
98 Refusal to authorise a clinical trial for ethical or safety reasons; Partial or complete clinical trial suspension or early termination of an ongoing clinical trial because of safety findings or lack of efficacy (see Section 3.15); Recall of investigational drug or comparator; Failure to obtain marketing approval for a tested indication including voluntary withdrawal of a marketing application; Risk management activities, including: Protocol modifications due to safety or efficacy concerns (e.g., dosage changes, changes in study inclusion/exclusion criteria, intensification of subject monitoring, limitation in trial duration); Restrictions in study population or indications; Changes to the informed consent document relating to safety issues; Formulation changes; Addition by regulators of a special safety-related reporting requirement Issuance of a communication to investigators or healthcare professionals Plans for new studies to address safety issues Significant Actions
; Recall of investigational drug or comparator; Failure to obtain marketing approval for a tested indication including voluntary withdrawal of a marketing application; Risk management activities, including: Protocol modifications due to safety or efficacy concerns (e.g., dosage changes, changes in study inclusion/exclusion criteria, intensification of subject monitoring, limitation in trial duration); Restrictions in study population or indications; Changes to the informed consent document relating to safety issues; Formulation changes; Addition by regulators of a special safety-related reporting requirement Issuance of a communication to investigators or healthcare professionals Plans for new studies to address safety issues Significant Actions")
99
99 Failure to obtain a marketing approval renewal Withdrawal or suspension of a marketing approval Significant restrictions on distribution or introduction of other risk minimisation measures Significant safety-related changes in labelling documents that could affect the development programme, including restrictions on use or population treated Communications to health care professionals New post-marketing study requirements imposed by regulators This section should also summarise requests from regulatory authority(ies) that place a specific limitation on current or future development (e.g., a request to conduct long-term animal studies before initiating a long-term clinical trial, specification of a maximum dose to be evaluated, a request for specific safety data before initiating trials in paediatric subjects). A cumulative listing of such requests from regulatory authorities should be provided, including any updates if applicable. This can be provided as a table, in an appendix, or in this section. Significant Actions related to marketed drugs

100
100 - Keine oder erheblich verspätete Einreichung von Sicherheitsberichten für Studien, die seit mehr als einem Jahr genehmigt sind und ein positives Ethikvotum haben - Nicht nachvollziehbare Beobachtungszeiträume - Unzureichende Angaben - Keine (S)AE-Meldungen trotz bereits erfolgtem Patienteneinschluss bei Indikationen oder IMPs, bei denen (S)AEs zu erwarten sind - Im Bericht nicht angegebene „Actions Taken for Safety Reasons“ Mögliche Trigger für GCP-Inspektion
AE-Meldungen trotz bereits erfolgtem Patienteneinschluss bei Indikationen oder IMPs, bei denen (S)AEs zu erwarten sind - Im Bericht nicht angegebene „Actions Taken for Safety Reasons Mögliche Trigger für GCP-Inspektion")
101
101 http://www.hma.eu/fileadmin/dateien/Human_Medicines/01- About_HMA/Working_Groups/CTFG/2011_12_22_Q___A_DSUR.pdf Ein nicht-kommerzieller Sponsor führt mehrere unabhängige klinische Studien mit einem IMP durch CTFG-Empfehlung: „single DSUR“ für das IMP erstellen Bei entsprechender Rechtfertigung kann auch ein „studienspezifischer DSUR“ akzeptiert werden Ein nicht-kommerzieller Sponsor führt eine klinische Studie mit einem noch nicht zugelassenen IMP durch CTFG-Empfehlung: Der DSUR für das IMP sollte vom zukünftigen Zulassungsinhaber (bzw. Hersteller) erstellt werden – Vereinbarung über Austausch von Sicherheitsdaten CTFG - Empfehlungen
 erstellt werden – Vereinbarung über Austausch von Sicherheitsdaten CTFG - Empfehlungen.")
102
102 1. Für alle Prüfpräparate außer Placebos übermittelt der Sponsor der AGENTUR jährlich über ein Modul der „EudraVigilance-Datenbank“ einen Bericht zur Sicherheit jedes Prüfpräparats, das in einer klinischen Prüfung, deren Sponsor er ist verwendet wird 2. Wenn bei einer klinischen Prüfung mehrere Prüfpräparate verwendet werden, kann der Sponsor, falls dies im Prüfplan vorgesehen ist, für alle bei dieser klinischen Prüfung eingesetzten Prüfpräparate einen einzigen Sicherheitsbericht übermitteln. 3. Der jährliche Bericht enthält nur zusammenfassende und anonymisierte Daten 4. Die Berichterstattung beginnt mit dem Tag der ursprünglichen Genehmigung für eine klinische Prüfung. Sie erlischt mit Ende der letzten klinischen Prüfung, die der Sponsor mit dem Prüfpräparat durchführt. Regulation 536/2014 Artikel 43 (ab Mai 2016) Jährliche Berichterstattung durch den Sponsor an die Agentur
 Jährliche Berichterstattung durch den Sponsor an die Agentur.")
103
103 clinicaltrials@ages.at corina.spreitzer@ages.at +43 (0) 505 55-36250 Vielen Dank für Ihre Aufmerksamkeit ! Bei Fragen zu DSUR / ASR:

104
Austrian Agency for Health and Food Safetywww.ages.at Häufige Fragen zur Klinischen Prüfung gemäß MPG Dr. Svetlana Seiter Institut Überwachung, Abteilung Klinische Prüfung svetlana.seiter@ages.at AGES-Gespräch Wien, 29.10.2015

105
105 Der Inhalt des Vortrags gibt die Sichtweisen der Vortragenden wieder und repräsentiert nicht offizielle Ansichten der AGES Medizinmarktaufsicht (MEA). Disclaimer

106
Übersicht Fragen zur Einstufung Fragen zu Kombistudien und Kombinationsprodukten Fragen bei Validierung und Begutachtung – Erstantrag Unterschiede im Marktzugang: AM vs. MP Ausblick in die Zukunft 106 Svetlana Seiter 2015-10- 29

107
Häufigste Fragen: Einstufung Ist das geplante Projekt eine Klinische Prüfung eines Medizin- produktes (MP)/Leistungsbewertungsprüfung eines IVD? Besteht eine Meldepflicht an das BASG? Kriterien: Medizinprodukt/IVD: vom Hersteller als MP/IVD ausgelobt oder durch den Einsatz in der Studie die Definition gemäß RL93/42/EWG bzw. RL98/79/EWG erfüllt § 3 MPG: Klinische Prüfung / Leistungsbewertungsprüfung - systematische Untersuchung eines Medizinproduktes / In-vitro- Diagnostikums…an Prüfungsteilnehmern / Proben…um Leistungsdaten, Nebenwirkungen, Risiken, Wirkungsmechanismen, geeignete klinische Einsatzgebiete…zu untersuchen 107 Svetlana Seiter 2015-10- 29

108
108 Svetlana Seiter 2015-10- 29 Gegenstand der Untersuchung des Projekts CE-Kennzeichen? Zusätzliche Dg/Th Maßnahmen? * Ja MP / IVD Anwendung in Zweckbestimmung? AIMD? MP-Studie, Meldepflicht an BASG MP-Studie, Meldepflicht an BASG Ja Nein *Gilt nicht für IVD Ja Nein Ja Nein Ja CE-Kennzeichen? Grundlagenforschung usw. MP im Projekt? Anwendung in Zweckbestimmung? Andere Rechtsmaterie Ja Nein CE-Kennzeichen? AM MP im Projekt? Anwendung in Zweckbestimmung? AM-Studie AM-MP- Kombistudie Ja Nein Ja Vergleich mit AM? Ja Nein

109
Wann wird ein Forschungsprojekt zur MP-Studie? Beispiele 1) Prüfprodukt - kein Medizinprodukt/IVD, durch die Zielsetzung in der KP ist die Definition eines MP/IVD‘s erfüllt Ergometer- vom Hersteller nicht als Medizinprodukt ausgelobt (Sportgerät) in der KP zum Zweck der Änderung von Durchblutungsparametern bei Patienten während eines bildgebenden Verfahrens systematisch untersucht =>Nicht-Medizinprodukt wird im Rahmen der KP zum Medizinprodukt Ziele des Projektes => maßgeblich dafür, ob MPG oder andere Rechtsmaterien als Grundlage für die KP-Durchführung herangezogen werden müssen 109 Svetlana Seiter 2015-10- 29
 Prüfprodukt - kein Medizinprodukt/IVD, durch die Zielsetzung in der KP ist die Definition eines MP/IVD‘s erfüllt Ergometer- vom Hersteller nicht als Medizinprodukt ausgelobt (Sportgerät) in der KP zum Zweck der Änderung von Durchblutungsparametern bei Patienten während eines bildgebenden Verfahrens systematisch untersucht =>Nicht-Medizinprodukt wird im Rahmen der KP zum Medizinprodukt Ziele des Projektes => maßgeblich dafür, ob MPG oder andere Rechtsmaterien als Grundlage für die KP-Durchführung herangezogen werden müssen 109 Svetlana Seiter")
110
Wann wird ein Forschungsprojekt zur MP-Studie? Beispiele 2) Medizinprodukte ohne CE-Kennzeichnung im Projekt Anwendung eines nicht CE-gekennzeichneten MP: z.B. ein radio- diagnostisches Gerätes (Eigenbau der Uni-Klinik) im Rahmen von „Grundlagenforschung“ eingesetzt => MP Studie Anwendung eines CE-gekennzeichneten, aber modifizierten MP (gilt auch für dessen Teile, Zubehör oder Software!) im Rahmen der KP = MP gilt als nicht CE-gekennzeichnet => MP Studie Anwendung eines CE-gekennzeichneten MP außerhalb seiner Zweckbestimmung = MP gilt als nicht CE-gekennzeichnet => MP Studie 110 Svetlana Seiter 2015-10- 29 Nicht CE-gekennzeichnete Medizinprodukte dürfen nur im Rahmen einer klinischen Prüfung angewendet werden (bis auf Ausnahmeregelungen) Sponsor übernimmt die Verantwortung des Herstellers in Bezug auf das MP
 Medizinprodukte ohne CE-Kennzeichnung im Projekt Anwendung eines nicht CE-gekennzeichneten MP: z.B. ein radio- diagnostisches Gerätes (Eigenbau der Uni-Klinik) im Rahmen von „Grundlagenforschung eingesetzt => MP Studie Anwendung eines CE-gekennzeichneten, aber modifizierten MP (gilt auch für dessen Teile, Zubehör oder Software!) im Rahmen der KP = MP gilt als nicht CE-gekennzeichnet => MP Studie Anwendung eines CE-gekennzeichneten MP außerhalb seiner Zweckbestimmung = MP gilt als nicht CE-gekennzeichnet => MP Studie 110 Svetlana Seiter Nicht CE-gekennzeichnete Medizinprodukte dürfen nur im Rahmen einer klinischen Prüfung angewendet werden (bis auf Ausnahmeregelungen) Sponsor übernimmt die Verantwortung des Herstellers in Bezug auf das MP.")
111
Wann wird ein Forschungsprojekt zur Kombinationsstudie? Beispiele 1) Arzneimittel-Studie, in der ein MP außerhalb der Zweckbestimmung zum Einsatz kommt Klinische Prüfung eines Chemotherapeutikums, welches mit einem CE- gekennzeichnetem MP verabreicht werden soll MP - für Verabreichung von Lösungen wie NaCl 0,9% oder Ringer indiziert, nicht für Chemotherapeutika evaluiert und validiert => Anwendung außerhalb der Zweckbestimmung => AM-MP Studie In diesem Fall nachgefordert: Ergebnisse der Risikoanalyse gemäß EN ISO 14971 Mögliche Reaktionen zwischen dem Chemotherapeutikum, dem Gerät und den Schlauchsystemen (z.B. Korrosion)? => eventuelle Gefährdungen für Patienten und Personal? 111 Svetlana Seiter 2015-10- 29
 Arzneimittel-Studie, in der ein MP außerhalb der Zweckbestimmung zum Einsatz kommt Klinische Prüfung eines Chemotherapeutikums, welches mit einem CE- gekennzeichnetem MP verabreicht werden soll MP - für Verabreichung von Lösungen wie NaCl 0,9% oder Ringer indiziert, nicht für Chemotherapeutika evaluiert und validiert => Anwendung außerhalb der Zweckbestimmung => AM-MP Studie In diesem Fall nachgefordert: Ergebnisse der Risikoanalyse gemäß EN ISO Mögliche Reaktionen zwischen dem Chemotherapeutikum, dem Gerät und den Schlauchsystemen (z.B. Korrosion). => eventuelle Gefährdungen für Patienten und Personal. 111 Svetlana Seiter")
112
Wann wird ein Forschungsprojekt zur Kombinationsstudie? Beispiele 2) Arzneimittel-Studie, in der ein MP außerhalb der Zweckbestimmung zum Einsatz kommt Klinische Prüfung eines Antiasthmatikums, das per Inhaler (wiederverwendbar) Kindern verabreicht werden soll MP trägt CE-Kennzeichnung Anwendung in der Klinischen Prüfung gemäß der Indikation (= Asthma) MP - nicht für die Population der Kinder ausgelobt => außerhalb der Zweckbestimmung, systematische Untersuchung des MP im neuen Anwendungsbereich => AM-MP Studie 112 Svetlana Seiter 2015-10- 29
 Arzneimittel-Studie, in der ein MP außerhalb der Zweckbestimmung zum Einsatz kommt Klinische Prüfung eines Antiasthmatikums, das per Inhaler (wiederverwendbar) Kindern verabreicht werden soll MP trägt CE-Kennzeichnung Anwendung in der Klinischen Prüfung gemäß der Indikation (= Asthma) MP - nicht für die Population der Kinder ausgelobt => außerhalb der Zweckbestimmung, systematische Untersuchung des MP im neuen Anwendungsbereich => AM-MP Studie 112 Svetlana Seiter")
113
Kombinationsprodukte 113 Svetlana Seiter 2015-10- 29 MEDDEV 2.1/3 rev.3: Borderline products, drug-delivery products and medical devices incorporating, as integral part, an ancillary medicinal substance or an ancillary human blood derivative, December 2009 RL 2001/83/EG: In Zweifelsfällen…ein Erzeugnis…sowohl unter die Definition von „Arzneimittel“ als auch unter die Definition eines Erzeugnisses fallen kann, das durch andere gemeinschaftliche Rechtsvorschriften geregelt ist, gilt diese Richtlinie. Hauptwirkmechanismus ausschlaggebend für Abgrenzung RL 2001/83/EG AM mit Medizinprodukt als integralem Teil (feste Kombination, MP nicht wiederverwendbar) Kombiniertes ATMP RL 93/42/EWG, RL 90/385/EWG, RL 98/79/EG MP enthält Arzneimittelkomponenten in unterstützender Funktion IVD: Companion diagnostics
 Kombiniertes ATMP RL 93/42/EWG, RL 90/385/EWG, RL 98/79/EG MP enthält Arzneimittelkomponenten in unterstützender Funktion IVD: Companion diagnostics.")
114
Aus den Nachforderungen zum Erstantrag… Formale Vollständigkeit Positive Stellungnahme der Ethikkommission Versicherungsbestätigung (AT-Recht) CE-Nachweise (Konformitätserklärung des Herstellers, NB-Zertifikat(e)) Gebrauchsanweisung des MP Inhaltliche Begutachtung MP (v.a. ohne CE): Beschreibung des MP (Teile, Materialien, Funktion) Risikoanalyse, Biokomp. usw. Evtl. Literaturreferenz - auf Anwendbarkeit prüfen; Zusammenfassung des Sponsors Design und Typ der Studie: Auswahl mit Hinsicht auf Zielsetzung (z.B. einarmig, offen, kleine Probandenanzahl) Besonders schützenswerte Personen – Einschluss begründen 114 Svetlana Seiter 2015-10- 29
: Beschreibung des MP (Teile, Materialien, Funktion) Risikoanalyse, Biokomp. usw. Evtl. Literaturreferenz - auf Anwendbarkeit prüfen; Zusammenfassung des Sponsors Design und Typ der Studie: Auswahl mit Hinsicht auf Zielsetzung (z.B. einarmig, offen, kleine Probandenanzahl) Besonders schützenswerte Personen – Einschluss begründen 114 Svetlana Seiter")
115
Zulassung / In-Verkehr Bringen 115 Svetlana Seiter 2015-10- 29 Pharmakovigilanz/ Medizinprodukte- Vigilanz nationale Behörden nat. Behörden, EMA AMMP nationale Behörden Klinische Prüfung nationale Behörden Hersteller (+) Benannte Stelle (NB) - frei wählbar Einmalige Marktzulassung: CE-Kennzeichen im ganzen EWR gültig Zentrales Register aller MPs derzeit nicht vorhanden EUDAMED – derzeit nicht öffentlich Gebrauchsanweisung (IFU): nicht zwingend öffentlich verfügbar (Pflicht: Hersteller legt IFU dem MP bei) Zulassung/ In-Verkehr Bringen nat. Behörden, EMA Gültigkeit der Zulassung je nach Verfahren (nat., DCP, MRP, CP) Zulassungsinformation: z.B. Austria-Codex, EMA- Website Fachinformation: öffentlich verfügbar z.B. Klasse I Produkte außer Im, Is; allg. IVDs
 Benannte Stelle (NB) - frei wählbar Einmalige Marktzulassung: CE-Kennzeichen im ganzen EWR gültig Zentrales Register aller MPs derzeit nicht vorhanden EUDAMED – derzeit nicht öffentlich Gebrauchsanweisung (IFU): nicht zwingend öffentlich verfügbar (Pflicht: Hersteller legt IFU dem MP bei) Zulassung/ In-Verkehr Bringen nat. Behörden, EMA Gültigkeit der Zulassung je nach Verfahren (nat., DCP, MRP, CP) Zulassungsinformation: z.B. Austria-Codex, EMA- Website Fachinformation: öffentlich verfügbar z.B. Klasse I Produkte außer Im, Is; allg. IVDs.")
116
Revision der MP-Richtlinien http://ec.europa.eu/growth/sectors/medical-devices/regulatory- framework/revision/index_en.htm Überarbeitung der MEDDEV Guidelines, z.B. MEDDEV 2.7/2… RL90/385/EWG Aktive implantierbare Medizinprodukte Rechtliche Basis in Überarbeitung… 116 Svetlana Seiter 2015-10- 29 RL93/42/EWG Medizinprodukte RL98/79/EG In-vitro-Diagnostika EU Verordnung Medizinprodukte EU Verordnung In-vitro-Diagnostika u.a. Verbesserung der NB-Qualifikation, der klinischen Evaluierung /Prüfung, Transparenz u.a. verbesserte klinische Bewertung, Personalisierte Medizin, Software, Kooperation EMA/NBs/nat. Behörden Unmittelbar rechtsgültig – ohne nat. Implementierung Inkrafttreten 2017-2019?

117
IVD in der Personalisierten Medizin / in AM Studien IVDs als Begleitdiagnostika (Companion diagnostics): Derzeit: Entwicklung und In-Verkehr Bringen regulatorisch von AM getrennt (parallele Verfahren für AM und IVD) Zukünftig: engere Zusammenarbeit von NB/nat. Behörden, EMA geplant oft IVD In-House-Tests (in einer Gesundheitseinrichtung sowohl hergestellte als auch verwendete Produkte) Auf der EU-Ebene nicht geregelt – RL 98/79/EG nicht anwendbar Nationale Bestimmungen, in AT - Verordnung für Konformitätsbewertung von Medizinprodukten => kein Verfahren für Konformitätsbewertung keine CE-Zertifizierung aber… Grundlegende Anforderungen müssen erfüllt sein ! (wissenschaftliche Dokumentation) © 2002 Creatas 117 Svetlana Seiter 2015-10- 29
 Auf der EU-Ebene nicht geregelt – RL 98/79/EG nicht anwendbar Nationale Bestimmungen, in AT - Verordnung für Konformitätsbewertung von Medizinprodukten => kein Verfahren für Konformitätsbewertung keine CE-Zertifizierung aber… Grundlegende Anforderungen müssen erfüllt sein . (wissenschaftliche Dokumentation) © 2002 Creatas 117 Svetlana Seiter")
118
Kurz zusammengefasst… Anwendung von MPs im Forschungsprojekt – evtl. eine MP-Studie? (Nicht CE-gekennzeichnetes MP nur im Rahmen einer klinischen Prüfung - bis auf Ausnahmeregelungen) Kombinationsstudien – beide Rechtsmaterien berücksichtigen Zulassung + Fachinformation (AM) vs. In-Verkehr-Bringen + Gebrauchsanweisung (MP, IVD) Unterschiede Neue Verordnungen, neue Guidelines… 118 Svetlana Seiter 2015-10- 29
 Kombinationsstudien – beide Rechtsmaterien berücksichtigen Zulassung + Fachinformation (AM) vs. In-Verkehr-Bringen + Gebrauchsanweisung (MP, IVD) Unterschiede Neue Verordnungen, neue Guidelines… 118 Svetlana Seiter")
119
E-Mail: clinicaltrials@ages.atclinicaltrials@ages.at Web: www.basg.gv.atwww.basg.gv.at 119 Svetlana Seiter 2015-10- 29 Ich danke für Ihre Aufmerksamkeit!

120
Austrian Agency for Health and Food Safetywww.ages.at EudraCT Antragsformular Häufigste Fehler bei Studieneinreichungen Matthias Seidl Abteilung Klinische Prüfung, Institut Überwachung, AGES MEA AGES GESPRÄCH Wien, 29.10.2015

121
Einbringer Unterlagen in Papierform + Antragsformular XML & PDF Behörde Infomail formale Nachforderung (Telefon oder Mail) Nachreichung Formale Vollständigkeit (Mail) EudraCT DB XML Clinicaltrialsregister BASG Abstimmungen 121 Matthias Seidl 29.10.2015 Überblick Ersteinreichung
 Nachreichung Formale Vollständigkeit (Mail) EudraCT DB XML Clinicaltrialsregister BASG Abstimmungen 121 Matthias Seidl Überblick Ersteinreichung")
122
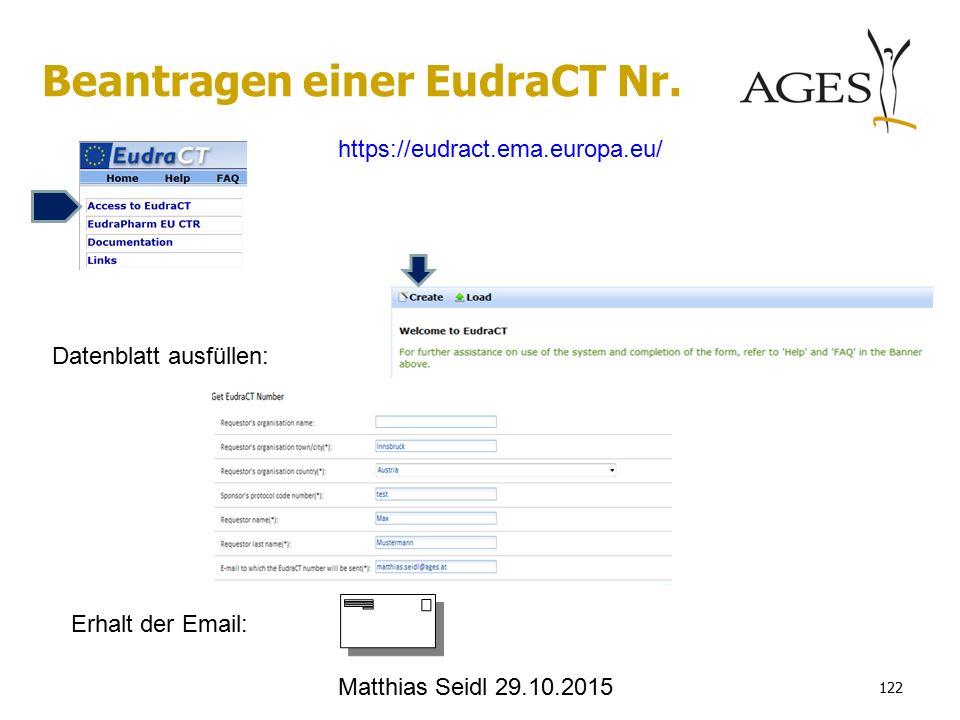
Datenblatt ausfüllen: Erhalt der Email: Matthias Seidl 29.10.2015 122 Beantragen einer EudraCT Nr. https://eudract.ema.europa.eu/
123
Matthias Seidl 29.10.2015 123 Erstellen des Antragsformular Auf der EudraCT Homepage können keine Daten an die Behörden übermittelt werden.

124
Matthias Seidl 29.10.2015 Achtung: Browsernavigation nicht verwenden Logout! 124 Übersicht

125
Auch bei einer Nachreichung oder Änderung ist First Submission auszuwählen. Resubmission A ist nur bei einer erneuten Einreichung nach Zurückziehung oder Ablehnung der Behörde auszuwählen. Matthias Seidl 29.10.2015 Bei multinationalen Studien (z.B. XML aus Deutschland) die zuständige Behörde ändern. Bitte auf das aktuelle Protokolldatum und Version achten! 125 ABSCHNITT A
 die zuständige Behörde ändern. Bitte auf das aktuelle Protokolldatum und Version achten. 125 ABSCHNITT A.")
126
Matthias Seidl 29.10.2015 Veröffentlichung: Kontaktdaten für Patienten für das öffentliche Register. Nicht für die Behörde! Kontaktdaten des Sponsors Wichtig für die Verrechnung. 126 ABSCHNITT B

127
Bitte auch die Abteilung eintragen. Matthias Seidl 29.10.2015 Kontaktdaten für Korrespondenz und Infomail. 127 ABSCHNITT C

128
ABSCHNITT D Steht das zugelassene Prüfpräparat zum Zeitpunkt der Einreichung fest, ist der Abschnitt D.2.2. mit „no“ zu beantworten. Bei zentral zugelassenen PR Prüfpräparat 1: Zugelassen, nicht modifiziert Matthias Seidl 2014-12-03 128

129
Die Dosis sollte nur bei einer „first in human“ Anwendung beantwortet werden. Matthias Seidl 29.10.2015 Ist nur bei zugelassenen nicht modifizierten Prüfpräparaten mit „yes“ zu beantworten. Prüfpräparat 1: Zugelassen, nicht modifiziert 129

130
Matthias Seidl 29.10.2015 Es sollte ausschließlich die Wirkstoffstärke und nicht die Dosis eingetragen werden. 130 Wirkstoffstärke

131
Falls im Rahmen dieser Studie verschiedene Wirkstoffstärken eines Arzneimittels getestet werden sollten alle Wirkstoffkonzentrationen als separate Prüfpräparate im Antragsformular eingetragen werden. Matthias Seidl 29.10.2015 131 Kopieren IMP

132
Prüfpräparat 2: Zugelassen, modifiziert Bei einem modifizierten Prüfpräparat ist eine Dokumentation von Ablauf und Kriterien der Herstellung des PRs im Rahmen eines Simplified IMPDs einzureichen. Matthias Seidl 29.10.2015 „Modified“ bedeutet Änderungen in der Herstellung, nicht der Indikation. 132

133
Matthias Seidl 29.10.2015 In diesem Fall sind nur sehr wenige Daten einzutragen. 133 Prüfpräparat 3: nicht Zugelassen, modifiziert

134
Matthias Seidl 29.10.2015 Die Substanz kann durch den Sponsor eingetragen werden. Die Substanz kann auch ohne Eintrag im EVMPD ausgefüllt werden. 134 Die Substanz ist noch nicht im EVMPD eingetragen: Die Substanz kann einmalig durch die EMA eingetragen werden.

135
Matthias Seidl 2014-12-03 135 Placebo ist identisch mit PR3:

136
Matthias Seidl 29.10.2015 Die Fachinformation der Kochsalzlösung ist als „Herstellungsdokumentation“ beizulegen. 136 Placebo nicht identisch (Natriumchlorid)
.")
137
Matthias Seidl 29.10.2015 137 Qualitätsfreigabe bei zugelassenem, nicht-modifzierten Prüfpräparat

138
Matthias Seidl 29.10.2015 138 Qualitätsfreigabe bei nicht zugelassenem oder modifiziertem Prüfpräparat:

139
Matthias Seidl 29.10.2015 ABSCHNITT E Muss mit den eingetragenen Prüfzentren in Abschnitt G. übereinstimmen. Oft nicht aktuell. 139

140
Matthias Seidl 29.10.2015 ABSCHNITT F Die Anzahl der Probanden muss übereinstimmen. Z.B. bei Demenz (§43 AMG) oder Koma (§43a AMG). Bei Kindern mit „no“ beantworten. 140
 oder Koma (§43a AMG). Bei Kindern mit „no beantworten")
141
Matthias Seidl 29.10.2015 ABSCHNITT G Gilt nicht als Prüfer im Sinne des AMG und zählt bei der Berechnung der Zentren nicht mit. Jedes Zentrum ist einmal einzutragen. Summe muss mit E.8.3 übereinstimmen. 141

142
Matthias Seidl 29.10.2015 ABSCHNITT H Die Einreichung bei der EK hat entweder vor oder zeitgleich mit der Einreichung beim BASG zu erfolgen 142

143
Matthias Seidl 29.10.2015 143 Speichern The Clinical Trial (EEA CTA) has passed all validation rules.
 has passed all validation rules.")
144
Matthias Seidl 29.10.2015 ABSCHNITT I 144

145
Vielen Dank für Ihre Aufmerksamkeit! 145 Matthias Seidl 29.10.2015 Fragen

Ähnliche Präsentationen



















