Präsentation herunterladen
Die Präsentation wird geladen. Bitte warten

1
Die Grundsätze der Pharmakologie

2
Einleitung (1) Pharmakodynamik ist die Lehre von den Wirkungen des Arzneimittels auf den Körper; dabei werden Fragen gestellt wie: Was macht das Arzneimittel mit dem Körper? Welche Rezeptoren aktiviert das Arzneimittel? Welche anderen Wirkungen hat das Arzneimittel? Arzneimittel wirken hauptsächlich auf zwei Arten: Sie verändern die Umgebung der Zellen. Beispiel: Antazida neutralisieren Säure im Magen Sie binden an Rezeptoren auf Zellmembranen und verändern so die Zellfunktion. Beispiel: Digoxin bindet an Rezeptoren auf Zellen des Herzens und verbessern dessen Fähigkeit, sich zusammenzuziehen Die Wechselwirkung eines Arzneimittels mit seinem Rezeptor ist mit dem Schlüssel-Schloss-Prinzip vergleichbar. Das Arzneimittel wirkt in einer von zwei Richtungen auf Rezeptoren: Agonist: das Arzneimittel stimuliert einen bestimmten Rezeptor durch seine Anbindung und erzeugt eine physiologische Wirkung. Beispiel: Noradrenalin stimuliert Rezeptoren auf Herzzellen, wodurch sich die Schlaggeschwindigkeit erhöht Antagonist: das Arzneimittel blockiert durch seine Anbindung den Rezeptor, so dass andere Stoffe ihn nicht mehr stimulieren können. Beispiel: das Antihistamin Diphenhydramin blockiert Histaminrezeptoren und verhindert so allergische Reaktionen

3
Einleitung (2) Pharmakokinetik (PK) ist die Lehre davon, wie der Körper mit dem Arzneimittel umgeht; dabei werden Fragen gestellt wie: Wie gelangt das Arzneimittel in den Körper? Wohin geht das Arzneimittel im Körper? Was macht der Körper mit dem Arzneimittel? Wie scheidet der Körper das Arzneimittel wieder aus? Mit PK-Daten wird untersucht, wie der Körper mit einem Arzneimittel umgeht. Solche Daten sind unverzichtbar für die Bestimmung von Dosishöhe und -häufigkeit. Mit einer Kombination naturwissenschaftlicher und mathematischer Modelle wird die Wanderung des Arzneimittels (und seiner Zwischen- und Abbauprodukte) durch den Körper im zeitlichen Verlauf erklärt und vorhergesagt. Viele pharmakokinetische Studien werden an Tieren durchgeführt (Toxikokinetik).
 durch den Körper im zeitlichen Verlauf erklärt und vorhergesagt. Viele pharmakokinetische Studien werden an Tieren durchgeführt (Toxikokinetik).")
4
In vorklinischen Studien wird das Sicherheitsprofil eines neuen Arzneimittels untersucht. Sie liefern wichtige Informationen darüber, ob der Wirkstoffkandidat in die erste Studie am Menschen (=klinische Studie) eintreten darf und in welcher Dosis. In jedem Lehrtext findet man zum Thema Pharmakokinetik das Kürzel ADME Absorption = wie das Arzneimittel im Körper aufgenommen wird Distribution = wohin das Arzneimittel im Körper verteilt wird Metabolismus = wie der Körper das Arzneimittel chemisch verändert (Stoffwechsel) Elimination = wie der Körper das Arzneimittel ausscheidet Diese vier Elemente sind zu berücksichtigen, wenn über die Pharmakologie eines Arzneimittels bei Menschen oder Tieren gesprochen wird. Zusammen liefern sie außerdem Daten über das Verhältnis zwischen den positiven und negativen (toxischen) Wirkungen. Mit diesen ADME-Elementen wird im Wesentlichen die Sicherheit/Verträglichkeit einer experimentellen Substanz beim Menschen vorhergesagt.
 Elimination = wie der Körper das Arzneimittel ausscheidet. Diese vier Elemente sind zu berücksichtigen, wenn über die Pharmakologie eines Arzneimittels bei Menschen oder Tieren gesprochen wird. Zusammen liefern sie außerdem Daten über das Verhältnis zwischen den positiven und negativen (toxischen) Wirkungen. Mit diesen ADME-Elementen wird im Wesentlichen die Sicherheit/Verträglichkeit einer experimentellen Substanz beim Menschen vorhergesagt.")
5
Grundsätze der Toxikokinetik
Ist definiert als die Ermittlung von PK-Daten an Tieren. Beschreibt die im Tierkörper erreichte Einwirkung der Substanz (Exposition) und deren Verhältnis zur Dosierung sowie die zeitlichen Abläufe. Konzentriert sich auf die Interpretation von Toxizitätstests. Es gibt keine festgelegten, detaillierten Empfehlungen für die Vorgehensweise in der Toxikokinetik. Ob toxikokinetische Daten gebraucht werden, wird von Fall zu Fall entschieden. Toxikokinetik ist definiert als die Gewinnung pharmakokinetischer Daten, entweder: integriert in die Durchführung vorklinischer Toxizitätsstudien oder durch Interpretation toxikologischer Ergebnisse und ihrer Bedeutung für klinische Sicherheitsaspekte. Das Hauptziel ist die Beschreibung der im Tierkörper erreichten Einwirkung der Substanz und deren Verhältnis zur Dosierung sowie die zeitlichen Abläufe. Aufgrund der Integration in Toxizitätstests und der Zwischenstellung zwischen vorklinischen und klinischen Studien liegt das Augenmerk hauptsächlich auf der Interpretation von Toxizitätstests und nicht auf der Bestimmung grundlegender pharmakokinetischer Parameter der geprüften Substanz. Da im dynamischen Prozess der Entwicklung eines Arzneimittels ein ständiger Informationsaustausch zwischen vorklinischen und klinischen Studien stattfindet, gibt es keine festgelegten, detaillierten Empfehlungen für die Vorgehensweise in der Toxikokinetik. Daher müssen nicht unbedingt in allen Studien toxikokinetische Daten erfasst werden. Wann solche Daten von Nutzen sind, ergibt sich aus dem wissenschaftlichen Urteilsvermögen. Der Bedarf an toxikokinetischen Daten und der Umfang der Expositionstests in einzelnen Toxizitätsstudien sollte flexibel Schritt für Schritt ermittelt werden, wobei von Fall zu Fall entschieden wird, ob die Informationen für eine Risiko- und Sicherheitsbewertung ausreichen.
 und deren Verhältnis zur Dosierung sowie die zeitlichen Abläufe. Konzentriert sich auf die Interpretation von Toxizitätstests. Es gibt keine festgelegten, detaillierten Empfehlungen für die Vorgehensweise in der Toxikokinetik. Ob toxikokinetische Daten gebraucht werden, wird von Fall zu Fall entschieden. Toxikokinetik ist definiert als die Gewinnung pharmakokinetischer Daten, entweder: integriert in die Durchführung vorklinischer Toxizitätsstudien oder. durch Interpretation toxikologischer Ergebnisse und ihrer Bedeutung für klinische Sicherheitsaspekte. Das Hauptziel ist die Beschreibung der im Tierkörper erreichten Einwirkung der Substanz und deren Verhältnis zur Dosierung sowie die zeitlichen Abläufe. Aufgrund der Integration in Toxizitätstests und der Zwischenstellung zwischen vorklinischen und klinischen Studien liegt das Augenmerk hauptsächlich auf der Interpretation von Toxizitätstests und nicht auf der Bestimmung grundlegender pharmakokinetischer Parameter der geprüften Substanz. Da im dynamischen Prozess der Entwicklung eines Arzneimittels ein ständiger Informationsaustausch zwischen vorklinischen und klinischen Studien stattfindet, gibt es keine festgelegten, detaillierten Empfehlungen für die Vorgehensweise in der Toxikokinetik. Daher müssen nicht unbedingt in allen Studien toxikokinetische Daten erfasst werden. Wann solche Daten von Nutzen sind, ergibt sich aus dem wissenschaftlichen Urteilsvermögen. Der Bedarf an toxikokinetischen Daten und der Umfang der Expositionstests in einzelnen Toxizitätsstudien sollte flexibel Schritt für Schritt ermittelt werden, wobei von Fall zu Fall entschieden wird, ob die Informationen für eine Risiko- und Sicherheitsbewertung ausreichen.")
6
Absorption (1) Das Wissen um die Vorgänge bei der Absorption ist von großer Bedeutung bei der Entwicklung pharmazeutischer Wirkstoffe. Die Absorption ist der Weg des Arzneimittels in die Blutbahn. Es gibt verschiedene Verabreichungswege. Auf dem nächsten Bild werden der orale und der intravenöse Verabreichungsweg hinsichtlich ihrer Bioverfügbarkeit verglichen – das ist die Geschwindigkeit, mit der der pharmazeutische Wirkstoff nach seiner Verabreichung biologisch verfügbar ist Absorption Dies ist der Weg des Arzneimittels in die Blutbahn. Es gibt verschiedene Verabreichungswege: 1. Orale Verabreichung (p.o.: per os – über den Mund) ist ein Verabreichungsweg, bei dem die Substanz durch den Mund eingenommen wird. Auch: peroral. 80 % der Arzneimittel in der medizinischen Praxis werden oral verabreicht praktisch und wirtschaftlich das Arzneimittel wird geschluckt über den Magen/Darm absorbiert gelangt über den Leberkreislauf in die Leber geht in den Körperkreislauf über 2. Sublingual (s.l.) das Arzneimittel löst sich unter der Zunge auf und wird durch die Schleimhäute in die Blutbahn aufgenommen Beispiel: Sublingualtabletten 3. Transdermal (t.d.) das Arzneimittel ist in einem Pflaster enthalten und wird durch die Haut aufgenommen Vorteil: die Dosierung erfolgt stetig und langfristig Nachteil: das Produkt ist teuer und kann durch den Kleber hautreizend wirken Beispiel: Nikotin-, Östrogenpflaster 4. Rektal (p.r.) das Arzneimittel liegt als Zäpfchen vor und wird in den Enddarm eingeführt, wo es durch die Schleimhäute in die Blutbahn aufgenommen wird Vorteil: kann bei Bewusstlosigkeit und Erbrechen angewendet werden Nachteil: unvollständige oder unregelmäßige Aufnahme ist möglich Beispiele: Zäpfchen gegen Schmerzen oder Übelkeit 5. Inhalativ das Arzneimittel wird als Gas, Aerosol oder Nebel eingeatmet wird meist angewendet, wenn das Mittel direkt auf das Lungengewebe wirken soll Vorteil: die Wirkung setzt schnell ein Nachteil: kann zur Reizung des Lungengewebes führen Beispiele: bronchialerweiternde Sprays 6. Intranasal das Arzneimittel wird durch die Nasenschleimhäute in die Blutbahn aufgenommen Beispiele: abschwellendes Nasenspray 7. Parenteral (intravenös - i.v., intramuskulär - i.m., subkutan - s.c) Vorteile: wirkt schneller als orale oder rektale Verabreichung umgeht die unberechenbare Absorption im Magen-Darm-Trakt nutzbar bei bewusstlosen Patienten Nachteile: muss steril angewendet werden, um Infektionen zu vermeiden teurer als andere Verabreichungswege schmerzt bei der Injektion
 ist ein Verabreichungsweg, bei dem die Substanz durch den Mund eingenommen wird. Auch: peroral. 80 % der Arzneimittel in der medizinischen Praxis werden oral verabreicht. praktisch und wirtschaftlich. das Arzneimittel wird geschluckt. über den Magen/Darm absorbiert. gelangt über den Leberkreislauf in die Leber. geht in den Körperkreislauf über. 2. Sublingual (s.l.) das Arzneimittel löst sich unter der Zunge auf und wird durch die Schleimhäute in die Blutbahn aufgenommen. Beispiel: Sublingualtabletten. 3. Transdermal (t.d.) das Arzneimittel ist in einem Pflaster enthalten und wird durch die Haut aufgenommen. Vorteil: die Dosierung erfolgt stetig und langfristig. Nachteil: das Produkt ist teuer und kann durch den Kleber hautreizend wirken. Beispiel: Nikotin-, Östrogenpflaster. 4. Rektal (p.r.) das Arzneimittel liegt als Zäpfchen vor und wird in den Enddarm eingeführt, wo es durch die Schleimhäute in die Blutbahn aufgenommen wird. Vorteil: kann bei Bewusstlosigkeit und Erbrechen angewendet werden. Nachteil: unvollständige oder unregelmäßige Aufnahme ist möglich. Beispiele: Zäpfchen gegen Schmerzen oder Übelkeit. 5. Inhalativ. das Arzneimittel wird als Gas, Aerosol oder Nebel eingeatmet. wird meist angewendet, wenn das Mittel direkt auf das Lungengewebe wirken soll. Vorteil: die Wirkung setzt schnell ein. Nachteil: kann zur Reizung des Lungengewebes führen. Beispiele: bronchialerweiternde Sprays. 6. Intranasal. das Arzneimittel wird durch die Nasenschleimhäute in die Blutbahn aufgenommen. Beispiele: abschwellendes Nasenspray. 7. Parenteral (intravenös - i.v., intramuskulär - i.m., subkutan - s.c) Vorteile: wirkt schneller als orale oder rektale Verabreichung. umgeht die unberechenbare Absorption im Magen-Darm-Trakt. nutzbar bei bewusstlosen Patienten. Nachteile: muss steril angewendet werden, um Infektionen zu vermeiden. teurer als andere Verabreichungswege. schmerzt bei der Injektion.")
7
Die Absorption (Übergang in den Blutkreislauf und Bioverfügbarkeit) ist je nach Verabreichungsweg unterschiedlich. Das Bild vergleicht die Absorption eines intravenös verabreichten Arzneimittel mit der eines oral verabreichten. Oral verabreichte Arzneimittel müssen verdaut werden, bevor der Wirkstoff in den Blutkreislauf gelangt (absorbiert wird). Bei intravenös verabreichten Arzneimitteln geschieht dies sofort.
. Bei intravenös verabreichten Arzneimitteln geschieht dies sofort..")
8
Distribution (1) Arzneimittel können auf vielen verschiedenen Wegen in den Körper gelangen. Im Zuge der Absorption gelangen sie vom Verabreichungsort in den Blutkreislauf. Distribution bezeichnet die Verteilung des Wirkstoffs im Körper. Der Grad der Distribution ist abhängig von den physikalischen und chemischen Eigenschaften des Wirkstoffs. B) Distribution Distribution bezeichnet die Verteilung des Wirkstoffs im Körper auf dem Weg zum vorgesehenen Ziel. Der Grad der Distribution eines Wirkstoffs hängt von seinen physikalischen und chemischen Eigenschaften ab sowie von seiner Fähigkeit, Zellmembranen, Blutgefäße, die Blut-Hirn-Schranke usw. zu durchdringen. Blut-Hirn-Schranke Nur fettlösliche Substanzen und sehr kleine Moleküle können diese dichten Zellverbindungen durchdringen, die den Blutstrom von der Hirnsubstanz trennen, und somit eine arzneiliche Wirkung entfalten Beispiel: Heroin wirkt stärker auf das Gehirn als Morphin, weil es besser fettlöslich ist Plasmaproteinbindung Viele Arzneimittel binden sich reversibel (d. h. zeitweise) an Plasmaproteine wie beispielsweise Albumin. Das Bindungsprofil zwischen Protein und Arzneimittel bestimmt also die Distribution und die Ausscheidungsrate. Nur ungebundene, also „freie“ Arzneimittel können durch Kapillarwände diffundieren eine arzneiliche Wirkung entfalten verstoffwechselt und ausgeschieden werden
 Distribution. Distribution bezeichnet die Verteilung des Wirkstoffs im Körper auf dem Weg zum vorgesehenen Ziel. Der Grad der Distribution eines Wirkstoffs hängt von seinen physikalischen und chemischen Eigenschaften ab sowie von seiner Fähigkeit, Zellmembranen, Blutgefäße, die Blut-Hirn-Schranke usw. zu durchdringen. Blut-Hirn-Schranke. Nur fettlösliche Substanzen und sehr kleine Moleküle können diese dichten Zellverbindungen durchdringen, die den Blutstrom von der Hirnsubstanz trennen, und somit eine arzneiliche Wirkung entfalten. Beispiel: Heroin wirkt stärker auf das Gehirn als Morphin, weil es besser fettlöslich ist. Plasmaproteinbindung. Viele Arzneimittel binden sich reversibel (d. h. zeitweise) an Plasmaproteine wie beispielsweise Albumin. Das Bindungsprofil zwischen Protein und Arzneimittel bestimmt also die Distribution und die Ausscheidungsrate. Nur ungebundene, also „freie Arzneimittel können. durch Kapillarwände diffundieren. eine arzneiliche Wirkung entfalten. verstoffwechselt und ausgeschieden werden.")
9
Nachdem ein Arzneimittel verabreicht wurde, verteilt es sich im Körper, bis es sein vorgesehenes Ziel erreicht.

10
Distribution (2) Einige der häufigsten Verabreichungswege von Arzneimitteln sind: oral (Einnehmen durch Schlucken), intramuskulär (Injektion in einen Muskel, z. B. im Arm), subkutan (Injektion dicht unter die Haut), intravenös (Verabreichung in eine Vene), oder transdermal (über ein Pflaster auf der Haut). Das Arzneimittel muss sein vorgesehenes Ziel erreichen.
, subkutan (Injektion dicht unter die Haut), intravenös (Verabreichung in eine Vene), oder transdermal (über ein Pflaster auf der Haut). Das Arzneimittel muss sein vorgesehenes Ziel erreichen.")
11
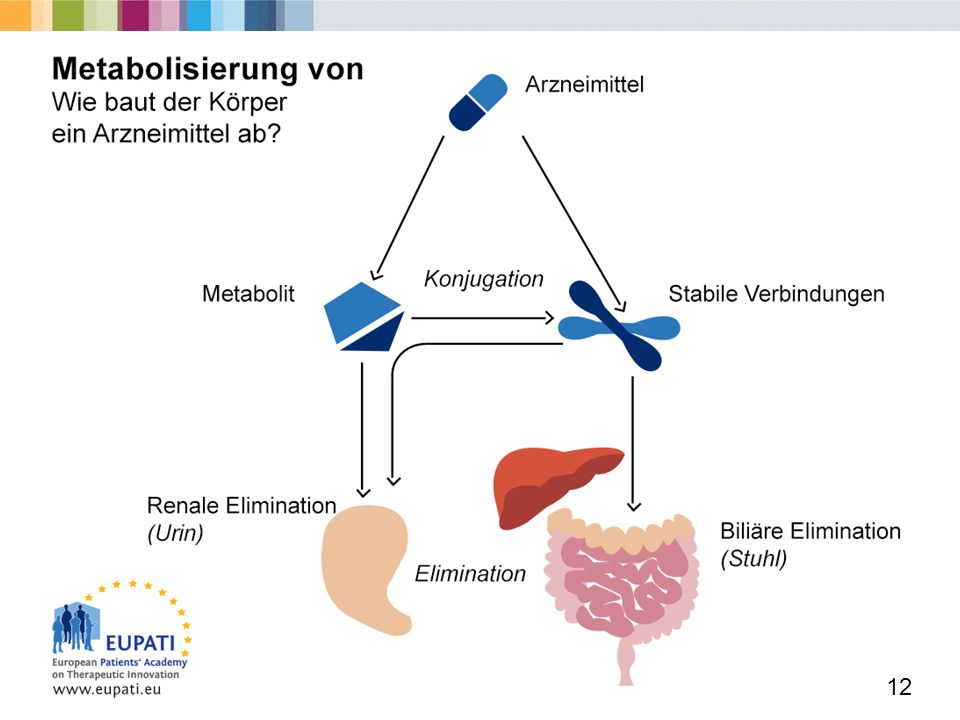
Metabolismus (1) Die Mehrzahl der Arzneimittel ist chemisch aktiv und wird im Körper verstoffwechselt. Das kann verschiedene Folgen haben: Verlust an Wirksamkeit Steigerung der Wirksamkeit Verringerung der Toxizität Steigerung der Toxizität C) Metabolismus (Biotransformation von Arzneimitteln) Metabolismus (auch Verstoffwechselung genannt) ist die Umwandlung einer chemischen Substanz in eine andere mithilfe von Enzymen. Ein Großteil des Metabolismus von Arzneimitteln findet in der Leber statt (im Bild mittig dargestellt), es gibt aber auch Vorgänge in der Darmwand, in der Lunge und im Blutplasma. Prinzipiell verändert sich durch Verstoffwechselung die therapeutische Wirkung von Arzneimitteln. Das komplexe Leberenzymsystem wandelt fettlösliche Arzneimittel in wasserlösliche Abbauprodukte um, die dann ausgeschieden werden können. „First-Pass-Effekt“ (Leber) Substanzen (Arzneimittel, Giftstoffe, Nahrung usw.), die aus dem Magen-Darm-Trakt in den Blutkreislauf übergehen, gelangen über die Pfortader in die Leber und dann in den großen Kreislauf. Der „First-Pass-Effekt“ bedeutet, dass die Leber Arzneimittel und mögliche Schadstoffe verstoffwechselt oder deaktiviert, bevor sie im ganzen Körper verteilt werden. Arzneimittel werden in der Leber in weniger aktive oder inaktive Zwischen- und Abbauprodukte umgewandelt. Die wichtigsten Arbeitszellen der Leber (die so genannten Hepatozyten) enthalten alle notwendigen Enzyme für die Verstoffwechselung von Arzneimitteln. Enzyme sind Proteine (Eiweiße), die die Geschwindigkeit chemischer Reaktionen im Körper bestimmen. Die wichtigsten am Metabolismus beteiligten Enzyme sind die der Cytochrom-P450-Gruppe. In der Phase 1 des Metabolismus können Arzneimittel einer Reduktion oder Hydrolyse unterworfen sein, die häufigste biochemische Reaktion ist aber die Oxidation. Oxidation ist die chemische Reaktion, die zum Beispiel auch beim Braunwerden von Äpfeln bei Kontakt mit Luftsauerstoff auftritt. Cytochrom-P450-Enzyme sind Katalysatoren der Oxidation. Bei dieser Reaktion verliert das Wirkstoffmolekül Elektronen (negativ geladene atomare Teilchen). Der Wirkstoff ist danach „oxidiert“. Der in Reaktionen der Phase 1 entstehende Metabolit ist oftmals immer noch chemisch aktiv. Ein Metabolit ist ein beim Metabolismus entstehendes Zwischenprodukt. Das können polare und unpolare Moleküle (oder Spezies) sein. In Phase 2 des Metabolismus kommt es zur Konjugation - also einer Verbindung des Wirkstoffs mit einer ionisierten Gruppe. Dadurch wird der Metabolit noch wasserlöslicher. Dies erleichtert die Ausscheidung (im Bild Elimination genannt) und senkt die arzneiliche Wirksamkeit. Renale Elimination (über die Nieren im Urin) Biliäre Elimination (über den Darm im Stuhl) Einige Arzneimittel-Zwischenprodukte können toxisch sein, zum Beispiel die von Paracetamol. Diese werden in der Phase 2 durch Konjugation mit Glutathion entgiftet. Glutathion ist das stärkste Antioxidant des Körpers. Es findet sich in jeder einzelnen Körperzelle. Falls jedoch eine Substanz, z. B. Paracetamol, zu hoch dosiert ist, gibt es nicht genug Glutathion, um die Zwischenprodukte zu entgiften. Sie sammeln sich an und wirken toxisch, wodurch es zu Leberschäden kommen kann. Als Lösung werden Substanzen beigegeben, die den Glutathionspiegel auffüllen, damit die Phase 2 des Metabolismus stattfinden kann, wodurch die Paracetamol-Zwischenprodukte vollständig entgiftet werden und die Leber geschont wird. University of Nottingham. RLO: The Liver and Drug Metabolism. (Stand:
 Metabolismus (Biotransformation von Arzneimitteln) Metabolismus (auch Verstoffwechselung genannt) ist die Umwandlung einer chemischen Substanz in eine andere mithilfe von Enzymen. Ein Großteil des Metabolismus von Arzneimitteln findet in der Leber statt (im Bild mittig dargestellt), es gibt aber auch Vorgänge in der Darmwand, in der Lunge und im Blutplasma. Prinzipiell verändert sich durch Verstoffwechselung die therapeutische Wirkung von Arzneimitteln. Das komplexe Leberenzymsystem wandelt fettlösliche Arzneimittel in wasserlösliche Abbauprodukte um, die dann ausgeschieden werden können. „First-Pass-Effekt (Leber) Substanzen (Arzneimittel, Giftstoffe, Nahrung usw.), die aus dem Magen-Darm-Trakt in den Blutkreislauf übergehen, gelangen über die Pfortader in die Leber und dann in den großen Kreislauf. Der „First-Pass-Effekt bedeutet, dass die Leber Arzneimittel und mögliche Schadstoffe verstoffwechselt oder deaktiviert, bevor sie im ganzen Körper verteilt werden. Arzneimittel werden in der Leber in weniger aktive oder inaktive Zwischen- und Abbauprodukte umgewandelt. Die wichtigsten Arbeitszellen der Leber (die so genannten Hepatozyten) enthalten alle notwendigen Enzyme für die Verstoffwechselung von Arzneimitteln. Enzyme sind Proteine (Eiweiße), die die Geschwindigkeit chemischer Reaktionen im Körper bestimmen. Die wichtigsten am Metabolismus beteiligten Enzyme sind die der Cytochrom-P450-Gruppe. In der Phase 1 des Metabolismus können Arzneimittel einer Reduktion oder Hydrolyse unterworfen sein, die häufigste biochemische Reaktion ist aber die Oxidation. Oxidation ist die chemische Reaktion, die zum Beispiel auch beim Braunwerden von Äpfeln bei Kontakt mit Luftsauerstoff auftritt. Cytochrom-P450-Enzyme sind Katalysatoren der Oxidation. Bei dieser Reaktion verliert das Wirkstoffmolekül Elektronen (negativ geladene atomare Teilchen). Der Wirkstoff ist danach „oxidiert . Der in Reaktionen der Phase 1 entstehende Metabolit ist oftmals immer noch chemisch aktiv. Ein Metabolit ist ein beim Metabolismus entstehendes Zwischenprodukt. Das können polare und unpolare Moleküle (oder Spezies) sein. In Phase 2 des Metabolismus kommt es zur Konjugation - also einer Verbindung des Wirkstoffs mit einer ionisierten Gruppe. Dadurch wird der Metabolit noch wasserlöslicher. Dies erleichtert die Ausscheidung (im Bild Elimination genannt) und senkt die arzneiliche Wirksamkeit. Renale Elimination (über die Nieren im Urin) Biliäre Elimination (über den Darm im Stuhl) Einige Arzneimittel-Zwischenprodukte können toxisch sein, zum Beispiel die von Paracetamol. Diese werden in der Phase 2 durch Konjugation mit Glutathion entgiftet. Glutathion ist das stärkste Antioxidant des Körpers. Es findet sich in jeder einzelnen Körperzelle. Falls jedoch eine Substanz, z. B. Paracetamol, zu hoch dosiert ist, gibt es nicht genug Glutathion, um die Zwischenprodukte zu entgiften. Sie sammeln sich an und wirken toxisch, wodurch es zu Leberschäden kommen kann. Als Lösung werden Substanzen beigegeben, die den Glutathionspiegel auffüllen, damit die Phase 2 des Metabolismus stattfinden kann, wodurch die Paracetamol-Zwischenprodukte vollständig entgiftet werden und die Leber geschont wird. University of Nottingham. RLO: The Liver and Drug Metabolism. (Stand:")
13
Elimination Arzneimittel und ihre Abbauprodukte können auf verschiedenen Wegen ausgeschieden werden. Dies sind, nach ihrer Bedeutung geordnet, folgende: über die Nieren im Stuhl abgeatmete Luft (über die Lungen) Schweiß (über die Haut) Von geringerer Bedeutung sind: Speichel Muttermilch D) Elimination Elimination bezieht sich auf die Ausscheidung eines Arzneimittels oder seiner Abbauprodukte aus dem Körper. Die drei wichtigsten Eliminationsrouten sind die Nieren, die Leber und der Darm. Die meisten Arzneimittel werden als aktive, teilweise inaktive oder vollständig inaktive Zwischen- bzw. Abbauprodukte über die Nieren ausgeschieden. 1) Elimination über die Nieren nach dem Metabolismus: Die Leber wandelt Arzneimittel in Stoffe um, die sich leichter über die Nieren ausscheiden lassen Elimination über die Nieren direkt in den Urin 2) Elimination im Stuhl Enterohepatischer Kreislauf: metabolisiertes Arzneimittel wird von der Leber in die Galle abgegeben und gelangt in den Dünndarm, von wo es erneut in den Blutkreislauf übergeht und in die Leber zurückkommt oder im Stuhl ausgeschieden wird Elimination von Arzneimittel und Lebensalter: Bei Säuglingen ist die Fähigkeit zur Metabolisierung und Elimination von Arzneimitteln noch unterentwickelt, wodurch sie empfindlicher sind Bei alten Menschen ist die Fähigkeit zur Metabolisierung und Elimination von Arzneimitteln beeinträchtigt, wodurch sie empfindlicher sind
 Schweiß (über die Haut) Von geringerer Bedeutung sind: Speichel. Muttermilch. D) Elimination. Elimination bezieht sich auf die Ausscheidung eines Arzneimittels oder seiner Abbauprodukte aus dem Körper. Die drei wichtigsten Eliminationsrouten sind die Nieren, die Leber und der Darm. Die meisten Arzneimittel werden als aktive, teilweise inaktive oder vollständig inaktive Zwischen- bzw. Abbauprodukte über die Nieren ausgeschieden. 1) Elimination über die Nieren nach dem Metabolismus: Die Leber wandelt Arzneimittel in Stoffe um, die sich leichter über die Nieren ausscheiden lassen. Elimination über die Nieren direkt in den Urin. 2) Elimination im Stuhl. Enterohepatischer Kreislauf: metabolisiertes Arzneimittel wird von der Leber in die Galle abgegeben und gelangt in den Dünndarm, von wo es erneut in den Blutkreislauf übergeht und in die Leber zurückkommt oder im Stuhl ausgeschieden wird. Elimination von Arzneimittel und Lebensalter: Bei Säuglingen ist die Fähigkeit zur Metabolisierung und Elimination von Arzneimitteln noch unterentwickelt, wodurch sie empfindlicher sind. Bei alten Menschen ist die Fähigkeit zur Metabolisierung und Elimination von Arzneimitteln beeinträchtigt, wodurch sie empfindlicher sind.")
14
Die vorklinische Phase
In vitro (Tier) In vitro (Mensch) In vivo Pharmakokinetik (Tier) Voraussagen für Erstanwendung beim Menschen In vivo Pharmakokinetik / Pharmakodynamik (Tier) Dieses Schaubild erläutert die Vorgehensweise bei der Forschung in der vorklinischen Phase der Arzneimittelentdeckung
 In vitro (Mensch) In vivo Pharmakokinetik (Tier) Voraussagen für Erstanwendung beim Menschen. In vivo Pharmakokinetik / Pharmakodynamik (Tier) Dieses Schaubild erläutert die Vorgehensweise bei der Forschung in der vorklinischen Phase der Arzneimittelentdeckung.")
15
Messwerte der Pharmakokinetik
Zuerst wird die Geschwindigkeit gemessen, mit der das Arzneimittel nach der Verabreichung im Blutkreislauf auftaucht (Absorption und Distribution). Dann misst man die Geschwindigkeit, mit der das Arzneimittel den Körper verlässt (Elimination) und wie es sich in der Zwischenzeit verändert (Metabolismus: Umwandlung in Zwischen- und Endprodukte, möglicherweise in mehreren Stufen. Dieses Schaubild zeigt das Bioverfügbarkeits-Profil eines intravenös verabreichten Arzneimittels. Hier sind wichtige Gesichtspunkte der Pharmakokinetik und Pharmakologie dargestellt: Y-Achse: Die Konzentration des Arzneimittels im Körper. X-Achse: Zeit. Wenn eine Person zum Zeitpunkt null ein Arzneimittel erhält, steigt die Konzentration des Arzneimittel an und sinkt dann langsam ab, bis das Arzneimittel ganz aus dem Körper ausgeschieden ist. Um das Bioverfügbarkeitsprofil zu bewerten und mit dem anderer Arzneimittel zu vergleichen, wird die so genannte „Fläche unter der Kurve“ (AUC, Area under the curve) herangezogen; diese gibt die so genannte Gesamtexposition des Körpers an, also das Ausmaß, in dem der Wirkstoff auf den Körper einwirkt.
. Dann misst man die Geschwindigkeit, mit der das Arzneimittel den Körper verlässt (Elimination) und wie es sich in der Zwischenzeit verändert (Metabolismus: Umwandlung in Zwischen- und Endprodukte, möglicherweise in mehreren Stufen. Dieses Schaubild zeigt das Bioverfügbarkeits-Profil eines intravenös verabreichten Arzneimittels. Hier sind wichtige Gesichtspunkte der Pharmakokinetik und Pharmakologie dargestellt: Y-Achse: Die Konzentration des Arzneimittels im Körper. X-Achse: Zeit. Wenn eine Person zum Zeitpunkt null ein Arzneimittel erhält, steigt die Konzentration des Arzneimittel an und sinkt dann langsam ab, bis das Arzneimittel ganz aus dem Körper ausgeschieden ist. Um das Bioverfügbarkeitsprofil zu bewerten und mit dem anderer Arzneimittel zu vergleichen, wird die so genannte „Fläche unter der Kurve (AUC, Area under the curve) herangezogen; diese gibt die so genannte Gesamtexposition des Körpers an, also das Ausmaß, in dem der Wirkstoff auf den Körper einwirkt.")
16
Das Arzneimittel gelangt schneller in den Körper hinein als wieder hinaus, weshalb die Konzentration des Arzneimittels im Körper ansteigt. Nachdem in dieser Kurve der höchste Punkt erreicht ist, wird das Arzneimittel schneller ausgeschieden als es noch aufgenommen wird, und damit sinkt die Konzentration wieder. Wichtig ist die gestrichelte rote Linie. Dies ist die Maximalkonzentration des Arzneimittels im Körper, abgekürzt Cmax. Wenn die Maximalkonzentration bekannt ist, lässt sich der Nutzen und auch die Möglichkeit von Nebenwirkungen besser vorhersagen. Die Zeit, bei der die Maximalkonzentration erreicht wird, heißt Tmax. Um das Bioverfügbarkeitsprofil zu bewerten und mit dem anderer Arzneimittel zu vergleichen, wird die so genannte „Fläche unter der Kurve“ (AUC, Area under the curve) herangezogen; diese gibt die so genannte Gesamtexposition des Körpers an, also das Ausmaß, in dem der Wirkstoff auf den Körper einwirkt.
 herangezogen; diese gibt die so genannte Gesamtexposition des Körpers an, also das Ausmaß, in dem der Wirkstoff auf den Körper einwirkt.")
17
Ein weiterer Begriff ist die Halbwertszeit eines Arzneimittels
Ein weiterer Begriff ist die Halbwertszeit eines Arzneimittels. Diese ist definiert als die Zeit, in der die aktuelle Konzentration des Arzneimittels im Körper um die Hälfte absinkt. In diesem Beispiel möchten wir wissen, wie lang es ab dem Zeitpunkt Tmax (hier 0) dauert, bis die Konzentration des Arzneimittel von Cmax (hier 10) auf die halbe Cmax (also 5) abgesunken ist. Die Zeit, innerhalb derer die Maximalkonzentration eines Arzneimittel auf die Hälfte absinkt, ist die Halbwertszeit, hier als lilafarbene Strecke dargestellt. Die Fläche unter der Kurve stellt die Gesamtexposition des Körpers gegenüber einem Arzneimittel dar. Sie ist der Ausdruck dessen, wie hoch die Konzentration des Arzneimittels ist und wie schnell es ausgeschieden wird. Um das Bioverfügbarkeitsprofil zu bewerten und mit dem anderer Arzneimittel zu vergleichen, wird die so genannte „Fläche unter der Kurve“ (AUC, Area under the curve) herangezogen; diese gibt die so genannte Gesamtexposition des Körpers an, also das Ausmaß, in dem der Wirkstoff auf den Körper einwirkt.
 dauert, bis die Konzentration des Arzneimittel von Cmax (hier 10) auf die halbe Cmax (also 5) abgesunken ist. Die Zeit, innerhalb derer die Maximalkonzentration eines Arzneimittel auf die Hälfte absinkt, ist die Halbwertszeit, hier als lilafarbene Strecke dargestellt. Die Fläche unter der Kurve stellt die Gesamtexposition des Körpers gegenüber einem Arzneimittel dar. Sie ist der Ausdruck dessen, wie hoch die Konzentration des Arzneimittels ist und wie schnell es ausgeschieden wird. Um das Bioverfügbarkeitsprofil zu bewerten und mit dem anderer Arzneimittel zu vergleichen, wird die so genannte „Fläche unter der Kurve (AUC, Area under the curve) herangezogen; diese gibt die so genannte Gesamtexposition des Körpers an, also das Ausmaß, in dem der Wirkstoff auf den Körper einwirkt.")
18
Arzneimittel und ADME Eine Leitstruktur-Verbindung ist noch kein Arzneimittel. Sie ist in der Regel der wirksamste entdeckte Wirkstoff. Damit die Leitstruktur-Verbindung ein wirksames Arzneimittel werden kann, müssen das pharmakologische Profil (einschließlich ADME-Faktoren), das toxikologische Profil, die Wirksamkeit und die Sicherheit zufriedenstellend sein.
, das toxikologische Profil, die Wirksamkeit und die Sicherheit zufriedenstellend sein.")
19
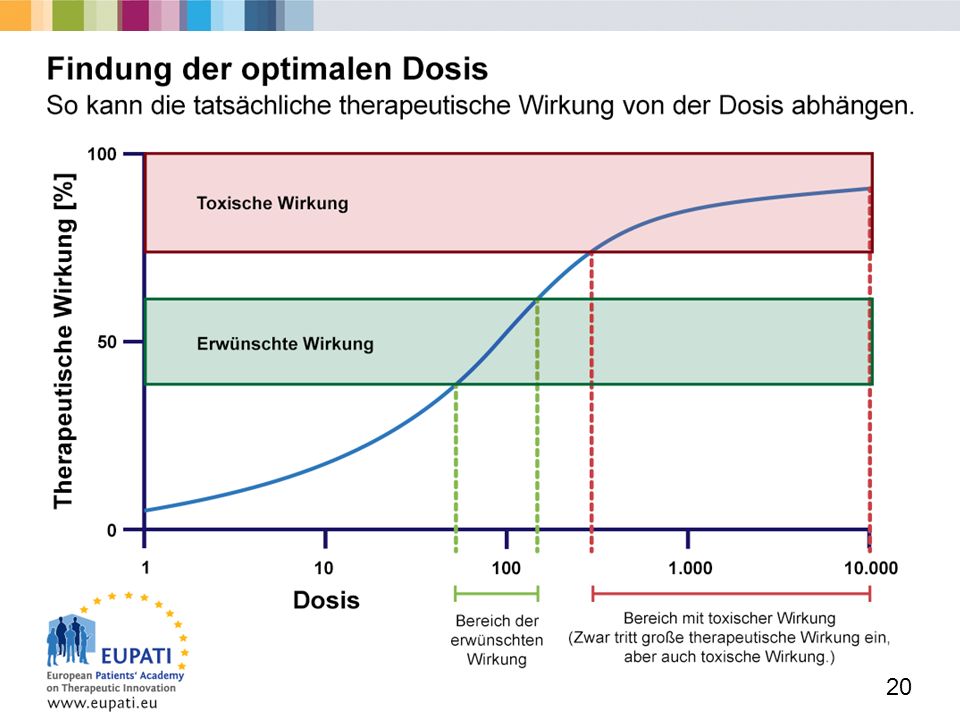
Die Startdosis für die klinische Entwicklung (1)
Eine sichere Startdosis für die Erstanwendung beim Menschen sollte durch Daten aus pharmakologischen und toxikologischen Studien an verschiedenen Tierarten gestützt sein, bevor mit Studien an Menschen begonnen wird. Aus toxikologischen Daten schließt man vor allem auf diejenige Startdosis, bei der die gewünschte Wirkung auftritt, ohne eine negative (toxische) Reaktion hervorzurufen. Aus den pharmakologischen Daten zieht man Rückschlüsse auf den Wirkmechanismus, die Beziehung zwischen Konzentration und Wirkung und andere Aspekte des pharmakokinetischen (PK) und pharmakodynamischen (PD) Profils.
 Reaktion hervorzurufen. Aus den pharmakologischen Daten zieht man Rückschlüsse auf den Wirkmechanismus, die Beziehung zwischen Konzentration und Wirkung und andere Aspekte des pharmakokinetischen (PK) und pharmakodynamischen (PD) Profils.")
21
Literatur: Guideline on Strategies to Identify and Mitigate Risks in First-in-Human Clinical Trials with investigational Medicinal Products e/2009/09/WC pdf Non-Clinical Safety Studies for the Conduct of Human Clinical Trials for Pharmaceuticals e/2009/09/WC pdf Preclinical safety evaluation of biotechnology-derived pharmaceuticals e/2009/09/WC pdf The Non-Clinical Evaluation of the Potential for delayed Ventricular Repolarisation (QT Interval Prolongation) by Human Pharmaceuticals Empfohlenes Video:
 by Human Pharmaceuticals Empfohlenes Video: v=mp93nPUzHqM&list=TLMdfpSMe8pGyVo0neC5BiZQ1xH6BQtidy.")
22
Literatur: Safety pharmacology studies for human pharmaceuticals e/2009/09/WC pdf Toxicokinetics: the assessment of systemic exposure in toxicology studies e/2009/09/WC pdf Position paper on the non-clinical safety studies to support clinical trials with a single micro dose e/2009/09/WC pdf Pharmacodynamics and Pharmacokinetics made ridiculously simple. Ezra Levy: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers

Ähnliche Präsentationen