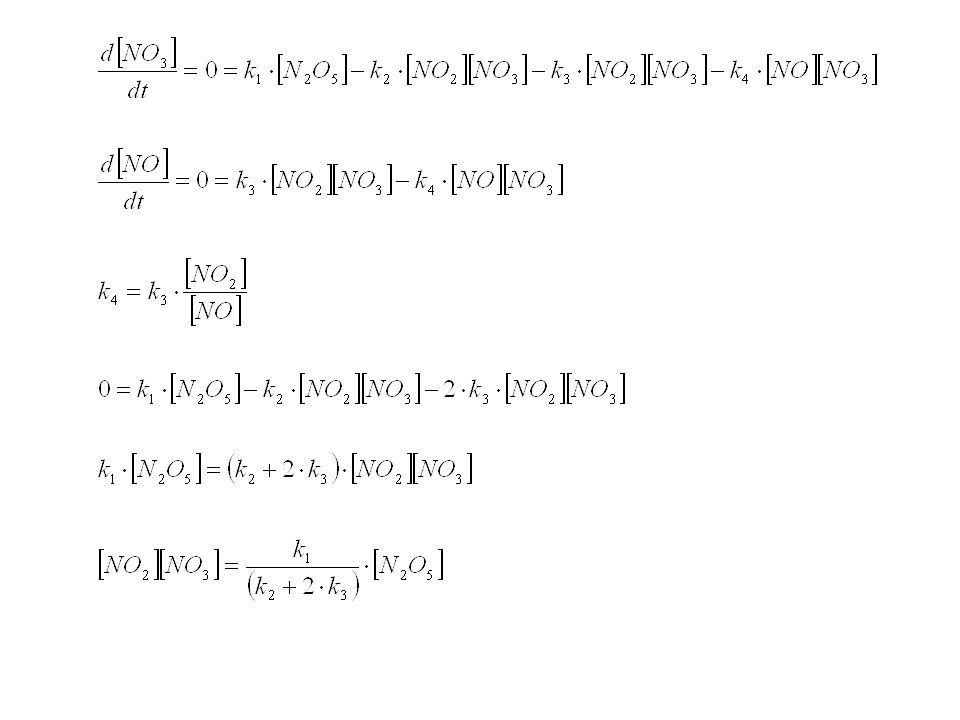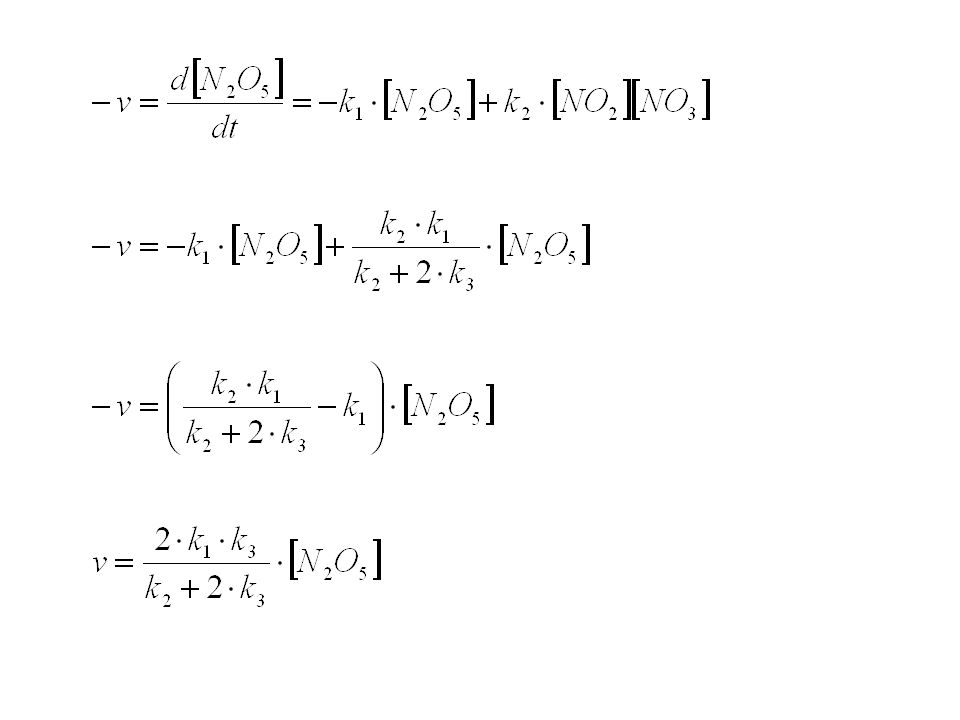Präsentation herunterladen
Die Präsentation wird geladen. Bitte warten

1
Beispiel: c c Quelle: R.G. Wilkins

2
Theoretische Berechnungen zur Polymerisation von Vinylacetat
W.H. Stockmayer, The Steady State Approximation in Polymerization Kinetics, J. Chem. Phys. 12 (4), 1944.
,")
3
Quasistationaritätsprinzip von Max Bodenstein (quasi-steady-state approximation):
Nach einer anfänglichen Induktionsphase bleibt die Konzentration der Zwischenverbindung über einen längeren Zeitabschnitt näherungsweise konstant.

4
“steady state“: Temperaturprofil ln(k/T) gegen 1/T NICHT linear!
 gegen 1/T NICHT linear!")
5
Druckabhängigkeit der Geschwindigkeitskonstante
Gibbs‘sche Fundamentalgleichung TST Eyring Aktivierungsvolumen lineares Druckprofil ln k gegen p

6
Aktivierungsvolumen Die Werte für können in der Nähe von Null liegen,
aber auch positiv oder negativ sein, mit Werten von einigen cm3/mol. Ist das Druckprofil nicht linear, so kann das zwei Gründe haben: Die Kompressibilität ist ungleich Null Die Geschwindigkeitskonstante k ist nicht einfach, sondern zusammengesetzt.

7
Chemische Interpretation der Aktivierungsparameter
Aktivierungsenthalpie: die Werte ergeben sich aus den Differenzen zwischen den Bindungenthalpien von Ausgangsstoffen und aktiviertem Komplex. Bei Reaktionen in Lösung kommen noch die Unterschiede in der Solvatisierungsenergie zwischen Ausgangsstoffen und aktiviertem Komplex hinzu.

8
Aktivierungsentropie:
Bimolekulare Reaktionen haben gewöhnlich negative Aktivierungsentropien, weil sich zwei Moleküle zu einem aktivierten Komplex zusammenfinden. Bei bimolekularen Gasreaktionen ist die Aktivierungsentropie immer negativ, denn der aktivierte Komplex besitzt immer weniger Freiheitsgrade als die Ausgangsmoleküle. Anders bei Reaktionen in Lösung, z.B. ist die Aktivierungsentropie positiv bei Aktivierter Komplex trägt geringere Ladung, Solvatation stark herabgesetzt Übergangszustand besitzt mehr Bewegungsfreiheiten als die Ausgangs-Ionen.

9
Es ist nicht einfach, genaue Messungen von zu machen.
Unimolekulare Reaktionen können Werte in der Nähe von 0 haben. Wenn bei der Reaktion Dissoziation erfolgt – und sich diese im aktivierten Komplex bereits abzeichnet – so ist positiv. Wechselwirkungen mit der Struktur des Lösungsmittels wirken sich stark aus: Zeichnet sich im aktivierten Komplex die Abdissoziation eines unpolaren Fragmentes ab, welches störend auf Struktur des umgebenden Wassers wirkt, so ist besonders stark positiv. Messwerte von geben also Hinweise auf die Struktur des Übergangszustandes. Es ist nicht einfach, genaue Messungen von zu machen. Man braucht dazu ein lineares Temperaturprofil über K.

10
Aktivierungsvolumen Maximale negative Werte: ca. – 20 cm3/mol Wenn während des Aktivierungsschrittes Ionen gebildet werden (= Volumenverlust des LM) „Normale“ negative Werte: ca. – 10 cm3/mol Wenn während des Aktivierungsschrittes Bindungen gebildet werden Positive Werte: ca. +10 cm3/mol Wenn sich im Übergangszustand das Aufbrechen von Bindungen abzeichnet (z.B. bei SN1 Reaktionen) Messwerte für sind gewöhnlich genauer als solche für Drucke in der Größenordnung 108 Pa nötig! und gehen, was Vorzeichen und Größe betrifft, in die gleiche Richtung.
 „Normale negative Werte: ca. – 10 cm3/mol. Wenn während des Aktivierungsschrittes Bindungen gebildet werden. Positive Werte: ca. +10 cm3/mol. Wenn sich im Übergangszustand das Aufbrechen von Bindungen abzeichnet (z.B. bei SN1 Reaktionen) Messwerte für sind gewöhnlich genauer als solche für. Drucke in der Größenordnung 108 Pa nötig! und gehen, was Vorzeichen und Größe betrifft, in die gleiche Richtung.")
11
Dimethylformamid DMF Dimethylsulfoxid DMSO Tetraethyldiethylentriamin et4dien Substitutionsreaktion eines quadratisch-planaren Komplexes

12
Theorie des Übergangszustandes (TST)
Lit.: H. Eyring, J. Chem. Phys. 1935, 3, Seite 107) Grundlegende Annahme: Quasi-Gleichgewicht zwischen den Reaktanden und dem Übergangszustand Die Bildung eines Übergangszustandes (= aktivierter Komplex) wird als charakteristisch für alle chemischen Änderungen angesehen.
 Grundlegende Annahme: Quasi-Gleichgewicht zwischen den Reaktanden und dem Übergangszustand. Die Bildung eines Übergangszustandes (= aktivierter Komplex) wird als charakteristisch für alle chemischen Änderungen angesehen.")
14
Die Aktivierungsparameter
sind die Freie Standard-Bildungsenthalpie, die Standard-Bildungsenthalpie und die Standard-Bildungsentropie des Aktivierten Komplexes

15
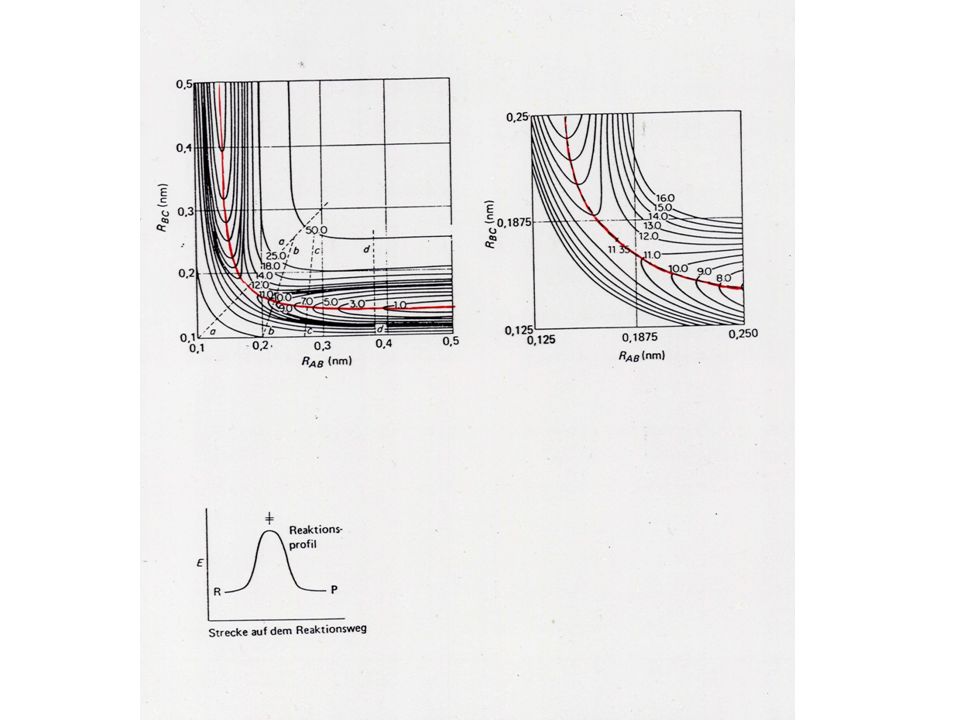
Energiediagramme= Reaktionsprofile Quelle: Moore/Hummel
Die potentielle Energie als Funktion einer Strecke auf dem Reaktionsweg für den molekularen Vorgang Reaktion läuft entlang der Reaktionskoordinate = Weg zwischen den Edukten und Produkten mit jeweils minimaler Änderung der potentiellen Energie. Sattelbereich der Potential-hyperfläche Quelle: Moore/Hummel

18
Ein System im Gleichgewicht:
Annahme: es gibt für diese Reaktion noch einen zweiten Reaktionsweg über die Verbindung B: Ist es möglich, ein Gleichgewicht zwischen A und C aufrecht zu erhalten unter der Bedingung, dass der Weg von A nach C auch über B führt, jedoch keine Rückreaktion von C nach B und von B nach A vorliegt? (=zyklisches Ausbalancieren der Reaktionsgeschwindigkeiten). NEIN! Durch ein allgemeines Prinzip aus der statistischen Mechanik streng verboten! k‘ k‘‘
. NEIN! Durch ein allgemeines Prinzip aus der statistischen Mechanik streng verboten! k‘ k‘‘")
19
Das Prinzip der mikroskopischen Reversibilität
NEIN!

20
Wenn eine Reaktion über mehrere Schritte und Wege erfolgt, so liegt im Gleichgewicht ein Netzwerk gekoppelter Gleichgewichte der Elementarreaktionen vor. Im Gleichgewicht gilt nicht nur für die Gesamtreaktion, dass die Geschwindigkeiten der Hin- und Rückreaktion gleich sein müssen, sondern es gilt dies auch für jede der beteiligten Elementarreaktionen.

21
Das Prinzip der mikroskopischen Reversibilität ist für einzelne molekulare Vorgänge gültig. Es kann aus den quantenmechanischen Ausdrücken für Übergangswahrscheinlichkeiten hergeleitet werden. Für jede Elementarreaktion gilt, dass der Übergangszustand der Hin- und Rückreaktion derselbe ist. Die Mechanismen der Hin- und Rückreaktion sind identisch: die Atome sitzen an denselben Stellen, der Unterschied ist nur, dass die Impulsvektoren in die entgegengesetzte Richtung weisen.

22
1. Ein Katalysator kann die Lage des Gleichgewichts nicht beeinflussen, da er die Geschwindigkeitskonstanten der Hin- und Rückreaktion um den gleichen Faktor verändert, beide Reaktionen verlaufen ja über denselben Übergangszustand. Könnte der Katalysator Hin- und Rückreaktion über unterschiedliche Übergangszustände führen, so könnte er die Gleichgewichtskonstante verändern. 2. Allgemeingültigkeit des MWG. Man kann jede Reaktion im Gleichgewicht aus gekoppelten Gleichgewichten ihrer Elementarreaktionen zusammensetzen. Man kann daher das MWG aus der Bruttoreaktionsgleichung direkt anschreiben, ohne den Reaktionsmechanismus, die beteiligten Elementarreaktionen und Zwischenverbindungen zu kennen.

23
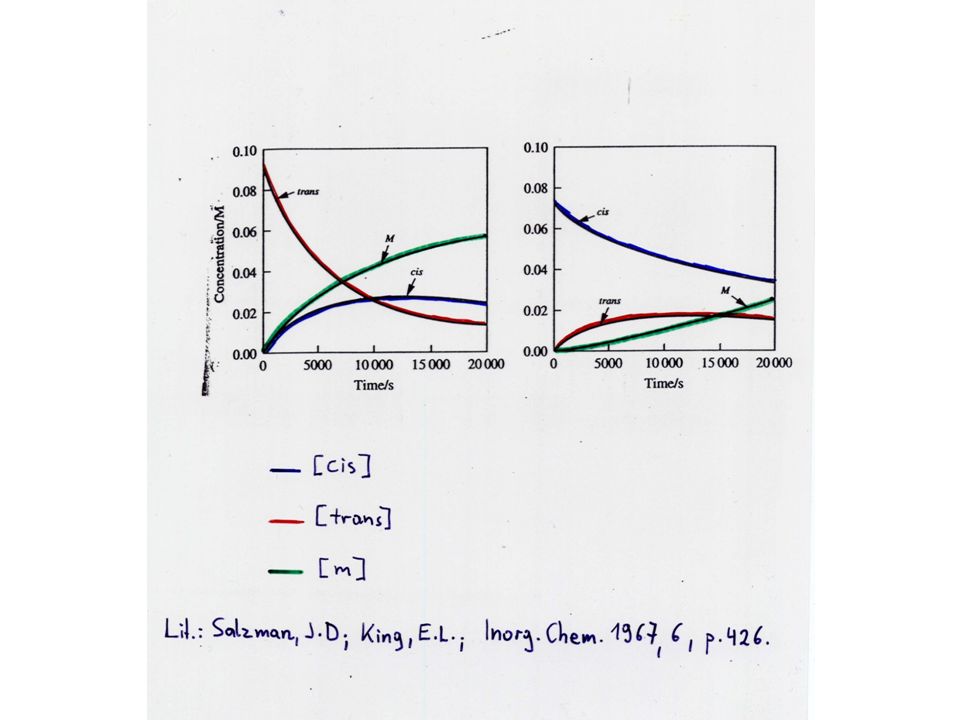
Beispiel: Isomerisierung und Verlust von Cl- von

24
Im Gleichgewicht gilt:
Es genügt fünf der sechs Geschwindigkeitskonstanten zu kennen.

26
Reaktionen in Lösung Bei Reaktionen in Lösung ist das Lösungsmittel am Übergangszustand beteiligt. Je nach Eigenschaften kann es den ÜZ stabilisieren oder destabilisieren. Sehr viele Reaktionen, die in Lösung stattfinden, sind in der Gasphase nicht möglich. Insbesondere gilt dies für ionische Reaktionen. Homolytische Zersetzungen von Neutralmolekülen können jedoch manchmal sowohl in der Gasphase als auch in der flüssigen Phase beobachtet werden: Beispiel a) Zersetzung von N2O5. Die Kinetik ist erster Ordnung, doch es handelt sich dabei NICHT um eine unimolekulare Reaktion. Die Geschwindigkeitskonstante dieser Reaktion ist fast dieselbe in der Gasphase und in verschiedenen Lösungsmitteln!
 Zersetzung von N2O5. Die Kinetik ist erster Ordnung, doch es handelt sich dabei NICHT um eine unimolekulare Reaktion. Die Geschwindigkeitskonstante dieser Reaktion ist fast dieselbe in der Gasphase und in verschiedenen Lösungsmitteln!")
30
Tetrachlormethan: unpolar
Nitromethan: polar

31
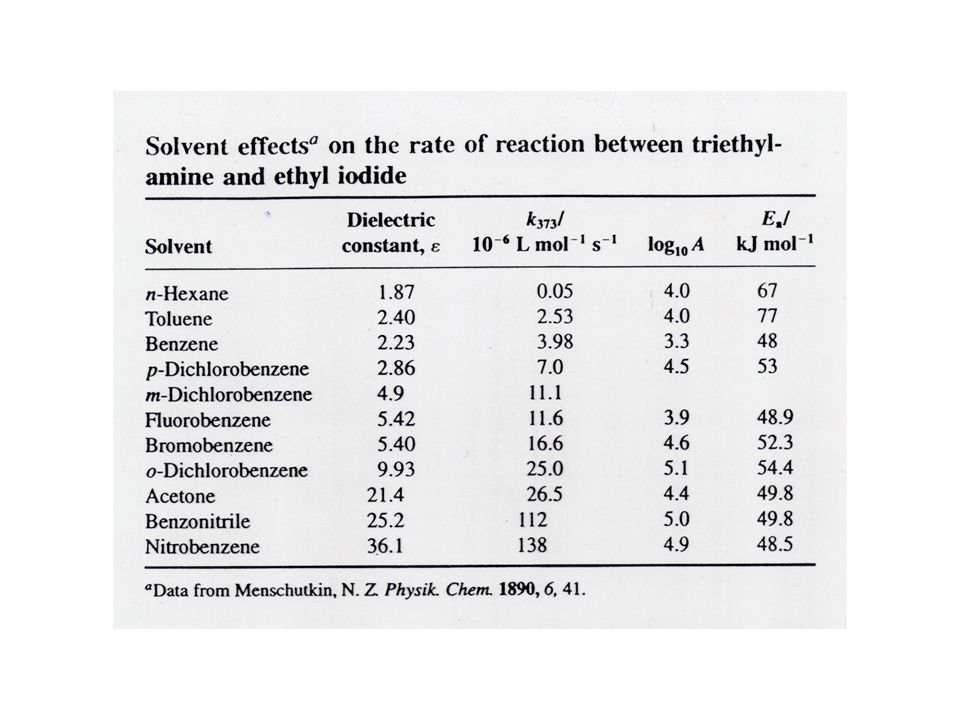
Beispiel b) Bildung von Tetraethylammoniumiodid Diese Reaktion gibt es in der Gasphase überhaupt nicht, aber sie kann in vielen unpolaren und polaren Lösungsmitteln nachgewiesen werden. Die Geschwindigkeitskonstante ist sehr stark vom Lösungsmittel abhängig. Von n-Hexan (unpolar) bis Nitrobenzol (polar) steigt die Geschwindigkeitskonstante um den Faktor 2700 Diese Reaktion hat einen polaren ÜZ der in LM mit hoher Permittivität (=dielektrische Leitfähigkeit, Dielektrizitätskonstante) stabilisiert wird.
 bis Nitrobenzol (polar) steigt die Geschwindigkeitskonstante um den Faktor Diese Reaktion hat einen polaren ÜZ der in LM mit hoher Permittivität (=dielektrische Leitfähigkeit, Dielektrizitätskonstante) stabilisiert wird.")
33
Das Molekül eines gelösten Stoffes befindet sich ständig in Wechselwirkung mit Molekülen des LM und muss über eine gewisse Strecke durch die Lösung diffundieren, bevor es auf ein anderes reaktionsfähiges Molekül trifft. Die Zahl solcher Zusammenstöße in der Zeiteinheit ist niedriger als in der Gasphase. Wenn sich zwei reaktionsfähige Moleküle aber erst einmal getroffen haben, dann bleiben sie ziemlich lange in unmittelbarer Nachbarschaft, umgeben von einem „Käfig“ aus LM-Molekülen. Innerhalb des Käfigs finden wiederholte Zusammenstöße zwischen ihnen statt (ca. 5-15) bevor sie sich wieder trennen. Die obere Grenze für die Geschwindigkeit einer bimolekularen Reaktion wird in Gasen durch die Stoßhäufigkeit gesetzt, in Flüssigkeiten durch die Häufigkeit der ersten Begegnung zwischen den reagierenden Molekülen, die sich in einer Brown‘schen Bewegung durch die Lösung bewegen.
 bevor sie sich wieder trennen. Die obere Grenze für die Geschwindigkeit einer bimolekularen Reaktion wird in Gasen durch die Stoßhäufigkeit gesetzt, in Flüssigkeiten durch die Häufigkeit der ersten Begegnung zwischen den reagierenden Molekülen, die sich in einer Brown‘schen Bewegung durch die Lösung bewegen.")
34
Es kann bei einer Reaktion in Lösung geschehen, dass die Teilchen, wenn sie erst einmal im Käfig sitzen, mit gegen 1 gehender Wahrscheinlichkeit miteinander reagieren. In einem solchen Fall ist die Diffusionsgeschwindigkeit geschwindigkeitsbestimmend für die Reaktion. Die Reaktion ist dann diffusionskontrolliert. Die Temperaturabhängigkeit einer diffusionskontrollierten Geschwindigkeitskonstante ist nur schwach und entspricht der Temperaturabhängigkeit des Diffusionskoeffizienten.

35
Reaktionen in Lösung langsam schnell
Geschwindigkeitskonstanten genauso interpretierbar wie bei Gasreaktionen Geschwindigkeitskonstanten abhängig von der Viskosität des Lösungsmittels, Reaktionen sind diffusionskontrolliert

Ähnliche Präsentationen