Präsentation herunterladen
Die Präsentation wird geladen. Bitte warten

1
Sequence Alignments --- Ilka Maria Axmann - Charité-Universitätsmedizin Berlin, Institut für Theoretische Biologie

2
introduction Was ist ein Alignment? Anordnung 2er oder mehrerer Sequenzen zur Untersuchung funktioneller, struktureller oder evolutionärer Verwandtschaften (Homologien) von Nukleotid- und Aminosäuresequenzen Wie sieht ein Alignment aus? gap gap match - konserviertes Element Seq1 HELIKS---TRN--HELIKS mismatch - Mutation Seq2 HELIKSSCHLEIFEHELIKS gap - Deletion oder Insertion (Indel) match mismatch match für lange/viele Sequenzen Computer notwendig Ähnlichkeit der einzelnen Elemente muss berechnet werden können -> Scoring Matrix (Bewertungsmatrix)
 von Nukleotid- und Aminosäuresequenzen Wie sieht ein Alignment aus. gap gap match - konserviertes Element Seq1 HELIKS---TRN--HELIKS mismatch - Mutation Seq2 HELIKSSCHLEIFEHELIKS gap - Deletion oder Insertion (Indel) match mismatch match für lange/viele Sequenzen Computer notwendig Ähnlichkeit der einzelnen Elemente muss berechnet werden können -> Scoring Matrix (Bewertungsmatrix).")
3
introduction Welche Arten, Algorithmen und Software gibt es? 2 Seq paarweises Alignment mehrere Seq multiples Alignment ( 1 Seq Rückfaltung, z.B. für RNA Seq) Lokales AlignmentFinden ähnlicher Teilsequenzen, z.B. Suche nach Seq.motiven, Proteindomänen Smith-Waterman-Algorithmus (klassisch) software: BLAST basic local alignment search tool (aktuell, heuristisch) Globales AlignmentAlle Elemente werden berücksichtigt, Sequenzen oft ähnlich lang und viele Seq.homologien zu erwarten Needleman-Wunsch-Algorithmus (dynamic programming, Laufzeit ~ Länge Anzahl ) software: CLUSTALW
 Lokales AlignmentFinden ähnlicher Teilsequenzen, z.B. Suche nach Seq.motiven, Proteindomänen Smith-Waterman-Algorithmus (klassisch) software: BLAST basic local alignment search tool (aktuell, heuristisch) Globales AlignmentAlle Elemente werden berücksichtigt, Sequenzen oft ähnlich lang und viele Seq.homologien zu erwarten Needleman-Wunsch-Algorithmus (dynamic programming, Laufzeit ~ Länge Anzahl ) software: CLUSTALW.")
4
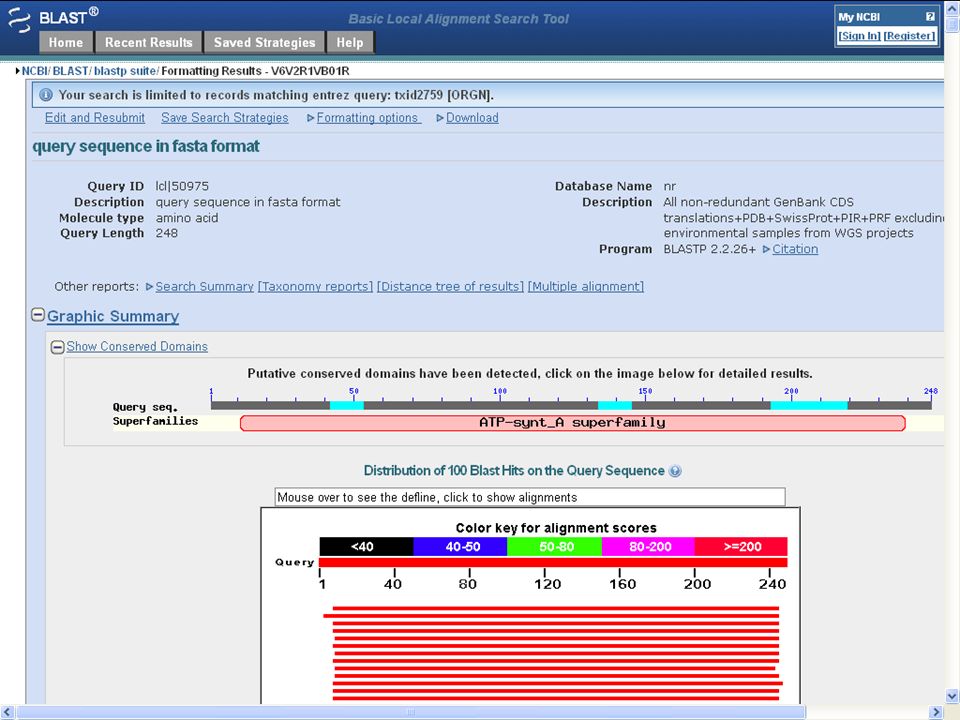
motivation: homology search NCBI/BLAST/blastp >query sequence in fasta format MLAGLSVLNLFPLAALEVGQQWYWEIGNLKLHGQTFATSWFVILLLVIASLAATRNVQRV PSGIQNLMEYVLEFLRDLARNQLGEKEYRPWLPFIGTLFLFIFVSNWSGALLPWKLIHIP DGAELAAPTNDINTTVALALLTSLAYFYAGLRKKGLGYFANYVQPIPVLLPIKILEDFTK PLSLSFRLFGNILADELVVAVLVLLVPLLVPLPLMALGLFTSAIQALVFATLAGAYIHEA IESEGEEH

6
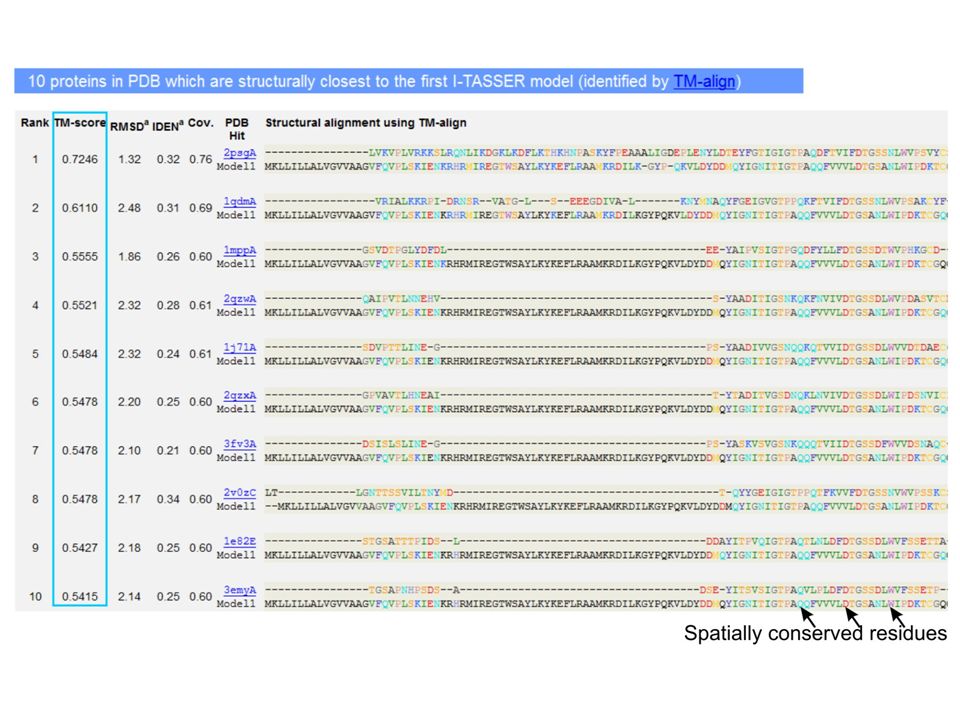
motivation: protein structure prediction Software zum Beispiel:I-TASSER (Zhang lab) Ambrish Roy, Dong Xu, Jonathan Poisson, Yang Zhang. A Protocol for Computer-Based Protein Structure and Function Prediction. Journal of Visualized Experiments, vol 57, e3259 (2011). Figure: An example of I-TASSER result page showing tertiary structure predictions for the query proteins.
. Figure: An example of I-TASSER result page showing tertiary structure predictions for the query proteins..")
8
motivation: phylogenetic trees Fig. © http://www.seas.upenn.edu/~cis120/hw/hw02/index.html

9
tree of life

10
motivation: similarity of nucleotide sequences

11
KaiC – protein phosphorylation ¤ conserved P-sites: S427 and T428 pS/pT S/pT or pS/T S/T SDS-PAGE & NanoLC-MS/MS - + SynKaiA 0 0.3 1 2 4 6 Hours at 18°C ProKaiC SynKaiC - + SynKaiA ¤ ProKaiC autophosphorylation without KaiA ¤ hypothesis: absence of kaiA compensated by enhanced autophosphorylation activity of KaiC A-loop (Kim et al., 2008) Fig. © Axmann et al., 2009, J Bact motivation: similarity/function of proteins

12
amino acids Carboxy- (-COOH) und Aminogruppe (-NH2) proteinogen (22) L-Aminosäuren kanonisch (20) Codon/Triplett (61) Fig. © Wikipedia

13
amino acids: physicochemical properties Carboxy- (-COOH) und Aminogruppe (-NH2) proteinogen (22) L-Aminosäuren kanonisch (20) Codon/Triplett (61) Fig. © Wikipedia

14
amino acids and codons # AS Codon 1 Start AUG 1 Met AUG 1 Trp UGG 2 Tyr UAU UAC 2 Phe UUU UUC 2 Cys UGU UGC 2 Asn AAU AAC 2 Asp GAU GAC 2 Gln CAA CAG 2 Glu GAA GAG 2 His CAU CAC 2 Lys AAA AAG 3 Ile AUU AUC AUA 4 Gly GGU GGC GGA GGG 4 Ala GCU GCC GCA GCG 4 Val GUU GUC GUA GUG 4 Thr ACU ACC ACA ACG 4 Pro CCU CCC CCA CCG 6 Leu CUU CUC CUA CUG UUA UUG 6 Ser UCU UCC UCA UCG AGU AGC 6 Arg CGU CGC CGA CGG AGA AGG 3 Stop UAA UAG UGA

15
substitution matrices 20 x 20 matrix PAM – point accepted mutations Margaret Dayhoff, 1970er Jahre Zahl substituierter Paare in nah verwandten Proteinen (Hämoglobin) PAM1: Rate der Substitution, wenn sich 1% der Aminosäuren verändern, entspricht Ähnlichkeit von 99% PAM250:höchste Stufe, Sequenzähnlichkeit von 20 % BLOSUM – BLOck Substitution Matrix Henikoff & Henikoff, 1992 Multiple Alignments evolutionär divergenter Proteine genutzt und darin Blöcke konservierter Sequenzabschnitte gefunden und ausgezählt BLOSUM62:am häufigsten genutzt, verwandte Proteinsequenz mit maximal 60% Identität
 PAM1: Rate der Substitution, wenn sich 1% der Aminosäuren verändern, entspricht Ähnlichkeit von 99% PAM250:höchste Stufe, Sequenzähnlichkeit von 20 % BLOSUM – BLOck Substitution Matrix Henikoff & Henikoff, 1992 Multiple Alignments evolutionär divergenter Proteine genutzt und darin Blöcke konservierter Sequenzabschnitte gefunden und ausgezählt BLOSUM62:am häufigsten genutzt, verwandte Proteinsequenz mit maximal 60% Identität")
16
substitution matrices: PAM250

17
substitution matrices: BLOSUM62

Ähnliche Präsentationen
>")

![Z-Transformation Die bilaterale Z-Transformation eines Signals x[n] ist die formale Reihe X(z): wobei n alle ganzen Zahlen durchläuft und z, im Allgemeinen,](/1/205073/big_thumb.jpg "Z-Transformation Die bilaterale Z-Transformation eines Signals x[n] ist die formale Reihe X(z): wobei n alle ganzen Zahlen durchläuft und z, im Allgemeinen,>")
















