Präsentation herunterladen
Die Präsentation wird geladen. Bitte warten

1
Softwarewerkzeuge der Bioinformatik
Inhalt dieser Veranstaltung: Softwarewerkzeuge für I Sequenzanalyse II Analyse von Proteinstruktur und Ligandenbindung III Zell- bzw. Netzwerksimulationen

2
„Lernziele“ Lerne aktuelle und bewährte Programme und Datenbanken der Bioinformatik kennen und erfolgreich einzusetzen um Tools kennenzulernen, mit denen man bioinformatische Fragen bearbeiten kann zu wissen, was auf dem Markt ist („das Rad nicht zweimal erfinden“) ein Gefühl dafür zu bekommen, wie erfolgreiche Softwareprodukte aussehen (sollen) 3 Mini-Forschungsprojekte zu bearbeiten Wir werden in der Vorlesung anhand von „Case-studies“ typische Fragestellungen in Pharma- oder Biotech-Unternehmen behandeln. Q: Wie stellen Sie sich den Arbeitsalltag als Bioinformatiker in einer Pharma-Firma vor?
 ein Gefühl dafür zu bekommen, wie erfolgreiche Softwareprodukte. aussehen (sollen) 3 Mini-Forschungsprojekte zu bearbeiten. Wir werden in der Vorlesung anhand von „Case-studies typische Fragestellungen in Pharma- oder Biotech-Unternehmen behandeln. Q: Wie stellen Sie sich den Arbeitsalltag als. Bioinformatiker in einer Pharma-Firma vor")
3
Organisatorisches Jede Woche zweistündige Vorlesung Freitag 9-11, Hörsaal 1, Geb. 45 Dozent: Prof. Helms Übungen „hands-on“ im CIP-Pool Bioinformatik Raum R 104 im Geb. 45 Freitag Uhr. Betreuer der Übungen Sequenz-Analyse Sam Ansari Proteinstruktur Dr. Michael Hutter Zellsimulationen Dr. Tihamer Geyer

4
Historische Entwicklung der Bioinformatik
1960‘er Jahre: Entwicklung phylogenetischer Methoden 1960‘er Jahre: Methoden zum Vergleich von DNA- und Proteinsequenzen 1976: erste MD-Simulation eines Proteins 1981: Smith-Waterman Algorithmus 1992: Sekundärstrukturvorhersage mit Neuronalen Netzwerken, PHD 1996: Vergleich von Proteinstrukturen mit DALI 2000: Durchbruch bei Sequenz-Assemblierung aus Shotgun-Daten (E. Myers)
")
5
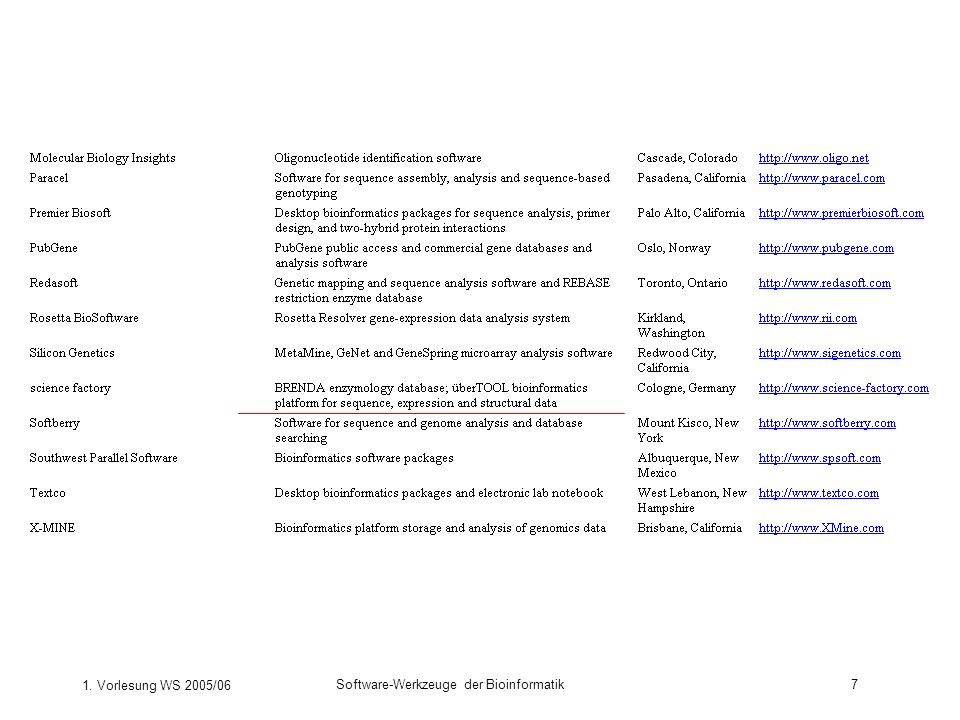
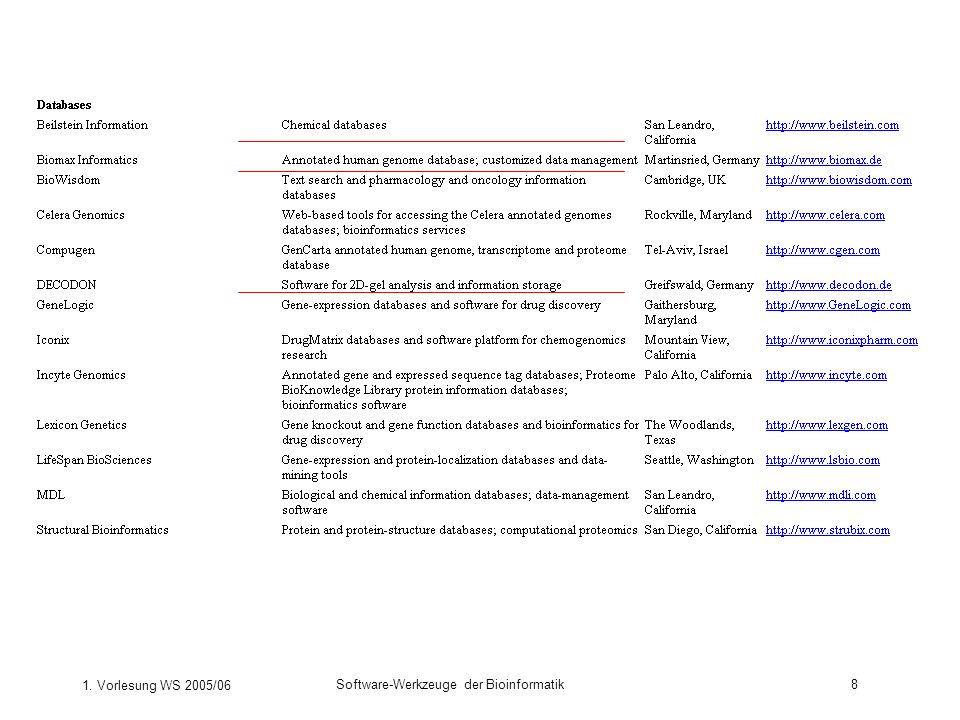
Welche Bioinformatik-Software gibt es?

12
Kommerzielle Software-Pakete sind bereits sehr mächtig
Kommerzielle Software ist sehr mächtig, da integriert, aber sehr teuer. Es ist fraglich, ob man in einer universitären Umgebung (mit kostenloser Software) bei Anwendungen im Bereich Drug Development mit Firmen konkurrieren kann, die solch mächtige Tools einsetzen.
 bei Anwendungen im Bereich Drug. Development mit Firmen konkurrieren. kann, die solch mächtige Tools einsetzen.")
13
Einsatz von Bioinformatik in der Produkt-Pipeline

14
Organisatorisches Jeder Teilnehmer an den Übungen benötigt einen Rechneraccount für den CIP-Pool. Diese Accounts werden von der Rechnerbetriebsgruppe des FB Informatik eingerichtet. Haben Sie bereits einen Account auf Uni-Rechnern? Dann muss dieser lediglich für den CIP-Pool freigeschaltet werden. Zugang zum CIP-Pool: Für Bioinformatik-Studenten 24/7, für alle anderen während der Übungsstunden. Bitte melden Sie sich nach dieser Stunde im Sekretariat des Zentrums für Bioinformatik bei Frau Alexandra Klasen an. Der Beginn der Übungen ist diese Woche.

15
Organisatorisches: Scheinvergabe
- Bewertung: Vorlesung zählt 2V + 2P = 9 Leistungspunkte Curriculum: Pflichtvorlesung für die Vertiefung „Bioinformatics“ (neue PO) kann für CMB-Bachelor eingebracht werden Wahlfach Pharmazie/Diplom, M.Sc. Biotechnologie - Benotung der Scheine: 50% der Benotung ergibt sich aus der mittleren Benotung von drei praktischen Aufgaben, die während des Semesters von jedem Studenten einzeln zu bearbeiten sind. Die Aufgaben werden etwa alle 4 Wochen ausgegeben und sind innerhalb von 2 Wochen zu bearbeiten und durch ein mindestens 5-seitiges Protokoll zu dokumentieren. Jeder Student muss mindestens zwei der drei praktischen Aufgaben mit einer Note von 4 und besser bestehen. Am Ende des Semesters wird eine 2-stündige Klausur über die Inhalte der Vorlesung und der Übungen geschrieben. Die Klausurnote geht ebenfalls mit 50% in die Scheinnote mit ein. Die Klausur muss mit einer Note von 4 und besser bestanden werden.
 kann für CMB-Bachelor eingebracht werden. Wahlfach Pharmazie/Diplom, M.Sc. Biotechnologie. - Benotung der Scheine: 50% der Benotung ergibt sich aus der mittleren Benotung von drei praktischen Aufgaben, die während des Semesters von jedem Studenten einzeln zu bearbeiten sind. Die Aufgaben werden etwa alle 4 Wochen ausgegeben und sind innerhalb von 2 Wochen zu bearbeiten und durch ein mindestens 5-seitiges Protokoll zu dokumentieren. Jeder Student muss mindestens zwei der drei praktischen Aufgaben mit einer Note von 4 und besser bestehen. Am Ende des Semesters wird eine 2-stündige Klausur über die Inhalte der Vorlesung und der Übungen geschrieben. Die Klausurnote geht ebenfalls mit 50% in die Scheinnote mit ein. Die Klausur muss mit einer Note von 4 und besser bestanden werden.")
16
Übersicht über Vorlesungsinhalt
I Sequenz Einführung Paarweises Sequenzalignment Multiples Sequenzalignment Phylogenie Techniken der Sequenzanalyse Genvorhersage, Transkriptionsfaktorbindungsstellen, Identifizierung von Repeats, CpG-Inseln, Assemblierung und Alignment von Genomen II Proteinstruktur Proteinstruktur Proteinstrukturvorhersage Proteinstruktur II Protein-Liganden-Wechselwirkung Protein-Protein-Docking III Zellsimulationen/Netzwerke E-Cell Virtual Cell Proteinnetzwerke metabolische Netzwerke

17
Was fange ich mit diesen Daten an?
Sequenz des menschlichen Genoms wurde 2001 entschlüsselt.

18
1 2 Analyse einer unbekannten Sequenz
Input: neue Proteinsequenz Experimentelle Daten vorhanden? Multiples Sequenzalignment Suche in Sequenzdatenbanken nach identischer Sequenz bzw. ähnlichen Sequenzen Erkenne Domänen Gibt es ähnliche Sequenz mit bekannter 3D-Struktur? Nein Vorhersage der Sekundärstruktur Zuordnung eines Protein-Folds Analyse dieses Folds, Nachbarn? Ja Ja Fold erkannt? Alignment der Sekundärstrukturen. Nein Modellierung der Proteinstruktur durch Homologiemodellierung Alignment der Sequenz mit einer Target-Struktur Ab inito Vorhersage der Tertiärstruktur 3D-Proteinstruktur Nach Rob Russell, gtsp/flowchart2.html Kann man Funktion zuordnen?

19
Sequenzanalyse 1

20
Ziele (0) Identifiziere alle menschlichen Proteine (ORFs) und ihre Funktion Sind dies alle Proteine? Nein: post-translationelle Modifikationen möglich wie Methylierung, Phosphorylierung, Glykosilierung … Identifiziere Gen-Netzwerke. Welche Proteine wechselwirken miteinander? (2) Identifiziere Module: abgeschlossene Einheiten (3) Identifiziere Sequenz-Abschnitte, in denen Mutationen für Krankheiten codieren
 Identifiziere Module: abgeschlossene Einheiten. (3) Identifiziere Sequenz-Abschnitte, in denen Mutationen für Krankheiten codieren.")
21
Sequenzen sind verwandt
Evolution findet auf vielen verschiedenen Ebenen statt : Mutationen einzelner Aminosäuren, Domänen-Shuffling, Genduplikation, Genom-Rearrangement verwandte Moleküle besitzen in verschiedenen Organismen ähnliche Funktionen (“Homologe”) Phylogenetischer Baum für ribosomale RNA: Drei Bereiche des Lebens
 Phylogenetischer Baum für ribosomale RNA: Drei Bereiche des Lebens.")
22
gap = Insertion oder Deletion
Sequenzalignment Der Zweck eines Sequenzalignments ist, all die Residuen einer beliebigen Anzahl von Sequenzen untereinander anzuordnen, die von der gleichen Residuenposition in einem Gen- oder Protein-Vorfahren abstammen. gap = Insertion oder Deletion Wat is het belangrijkste residue voor alignen? Cys, want meest geconserveerd A multiple sequence alignment is a 2D table, in which the rows represent individual sequences, and the columns the residue positions. Sequences are laid onto this grid in such a manner that (a) the relative positioning of residues within any one sequence is preserved, and (b) similar residues in all the sequences are brought into vertical register. Wie soll dies ein Computerprogramm entscheiden?
 the relative positioning of residues within any one sequence is preserved, and (b) similar residues in all the sequences are brought into vertical register. Wie soll dies ein Computerprogramm entscheiden")
23
Sequenzvergleiche: PAM250 Matrix
Für Sequenzvergleiche werden Bewertungsmatrizen für den Austausch von Aminosäuren verwendet. 1) Notice 1 lettercode for the amino acids on both axes are the 20 aa note blocks of similar amino acids 2) Symmetric, only one half shown 3) Diagonal: * For example: high score for matching Tryptophans and “low” score for matching Alanines. * Cysteine * Leu abundant 4) Off-diagonal Groups of similar amino acids K -> F -5 A score above zero assigned to two amino acids indicates that these two .. Each other more often than expected by chance alone. Ie they are functionall.. Exchangable A negative score indicates that the two amino acids are rarely .. Interchangeable. Eg. A basic amino acids for an acidic one or one with an … side chain for one with aliphatic side chain. Q: Warum sind manche Werte positiv, manche negativ? Q: Macht es Sinn, separate Austauschmatrizen für Membranproteine zu konstruieren?
 Notice 1 lettercode for the amino acids on both axes are the 20 aa note blocks of similar amino acids 2) Symmetric, only one half shown 3) Diagonal: * For example: high score for matching Tryptophans and low score for matching Alanines. * Cysteine. * Leu abundant. 4) Off-diagonal Groups of similar amino acids K -> F -5. A score above zero assigned to two amino acids indicates that these two .. Each other more often than expected by chance alone. Ie they are functionall.. Exchangable. A negative score indicates that the two amino acids are rarely .. Interchangeable. Eg. A basic amino acids for an acidic one or one with an … side chain for one with aliphatic side chain. Q: Warum sind manche Werte positiv, manche negativ Q: Macht es Sinn, separate Austauschmatrizen für Membranproteine zu konstruieren")
24
Proteinstruktur Sequenz
Was hat nun Sequenz-Konservierung mit Proteinstrukturen zu tun? sehr viel! Die Twilight zone kennzeichnet das Mass an Sequenzidentität, bis zu der zwei Proteinstrukturen mit hoher Wkt. die gleiche Struktur besitzen. Richtlinien von Doolittle: Sequenzen mit > 150 Residuen und 25% Sequenzidentität sind wahrscheinlich verwandt mit 15-20% Sequenzidentität können sie verwandt sein bei <15% Sequenzidentität ist es schwierig zu sagen ob sie verwandt sind oder nicht ohne weitere strukturelle oder funktionelle Hinweise TWILIGHT ZONE 1

25
Definition: Super-Sekundärstruktur
Die Anordnung (packing) von Sekundärstrukturelemente zu stabilen Einheiten wie b-barrels, bab Einheiten, Greek keys, usw.
 von Sekundärstrukturelemente zu stabilen Einheiten. wie b-barrels, bab Einheiten, Greek keys, usw.")
26
Definition: Tertiärstruktur
Die gesamte Faltung einer Kette, die sich aus der Packung der Sekundärstrukturelemente ergibt. Grün Fluoreszierendes Protein. Seine zylindrische Architektur wird durch 11 -Stränge gebildet. (1emb.pdb Brejc et al. 1997)
")
27
Einleitung: Proteinstruktur
cAMP-abhängige Proteinkinase Ca2+ Pumpe (katalytische Untereinheit) (TM Protein) 1
 (TM Protein) 1.")
28
Definition: Quartäre Struktur
Die Anordnung mehrerer Ketten eines Proteins, das mehrere Untereinheiten besitzt. Beispiel Hämoglobin

29
Proteinstruktur Sequenz
Konservierung von Residuen sind Indizien für den Verwandtschaftsgrad von Proteinen, für die Evolution und für die Verwandtschaft von Organismen Q: aus welchen Gründen können bestimmte Bereiche der Proteinsequenz konserviert sein? Konservierung von Residuen im aktiven Zentrum Konservierung von Residuen, die die Architektur der Proteinstruktur stabilisieren Konservierung von Residuen, die während Faltung des Proteins wichtig sind Konservierung von Residuen an Bindungsschnittstellen für Liganden und andere Proteine 1

30
Bedeutung von Sequenzanalyse
>900,000 Sequenzen in öffentlichen Datenbanken zugänglich Millionen mehr in proprietären dbs Anstieg wird mit Sequenzierung von weiteren Genomen weitergehen Was kann man diesen Informationen anfangen? In den Sequenzen steckt eine grosse Menge an strukturellen, funktionellen und evolutionären Informationen Sie sind eine sehr wichtige Datenquelle Im Gegensatz dazu gibt es nur etwa 2000 unabhängige Proteinstrukturen

31
Sequenz-Struktur Missverhältnis
800 700 600 500 400 300 200 100 Anzahl an nicht-redundanten Sequenzen ( ) Entsprechende Zunahme der Zahl an Proteinstrukturen ( ).
 Entsprechende Zunahme der Zahl an Proteinstrukturen ( ).")
32
Der “holy grail” der strukturellen Bioinformatik
Q: Wo stehen wir im Bereich der Strukturvorhersage? Homologiemodellierung Threading Ab initio Strukturvorhersage

33
Eigenschaften der Aminosäuren
Aminosäuren unterscheiden sich in ihren physikochemischen Eigenschaften. Q: müssen Bioinformatiker die Eigenschaften von Aminosäuren kennen?

34
Transmembrandomänen: Hydrophobizitätsskalen
TM Helices sind 20 Residuen lange Abschnitte aus vorwiegend hydrophoben Resiuden. Stephen White group, UC Irvine

35
Faltung von TM Proteinen wird durch Translokon unterstützt
Modell: Die neu synthesierte Polypeptidkette eines Membranproteins gelangt vom Ribosom durch den Translokon-komplex in die Membran (EM Abbildung). Erster Eindruck: Die Faltung eines Membranproteins ist ein hochkomplizierter Prozess. White, FEBS Lett. 555, 116 (2003)
. Erster Eindruck: Die Faltung eines Membranproteins ist ein. hochkomplizierter Prozess. White, FEBS Lett. 555, 116 (2003)")
36
Detektion der Membraninsertion via Glykosylierung
Integration of H-segments into the microsomal membrane a, Wild-type Lep has two N-terminal TM segments (TM1 and TM2) and a large luminal domain (P2). H-segments were inserted between residues 226 and 253 in the P2-domain. Glycosylation acceptor sites (G1 and G2) were placed in positions 96–98 and 258–260, flanking the H-segment. For H-segments that integrate into the membrane, only the G1 site is glycosylated (left), whereas both the G1 and G2 sites are glycosylated for H-segments that do not integrate in the membrane (right). b, Membrane integration of H-segments with the Leu/Ala composition 2L/17A, 3L/16A and 4L/15A. Bands of unglycosylated protein are indicated by a white dot; singly and doubly glycosylated proteins are indicated by one and two black dots, respectively. Hessa et al., Nature 433, 377 (2005)
 and a large luminal domain (P2). H-segments were inserted between residues 226 and 253 in the P2-domain. Glycosylation acceptor sites (G1 and G2) were placed in positions 96–98 and 258–260, flanking the H-segment. For H-segments that integrate into the membrane, only the G1 site is glycosylated (left), whereas both the G1 and G2 sites are glycosylated for H-segments that do not integrate in the membrane (right). b, Membrane integration of H-segments with the Leu/Ala composition 2L/17A, 3L/16A and 4L/15A. Bands of unglycosylated protein are indicated by a white dot; singly and doubly glycosylated proteins are indicated by one and two black dots, respectively. Hessa et al., Nature 433, 377 (2005)")
37
Translocon-assisted folding of TM proteins?
c, Gapp values for H-segments with 2–4 Leu residues. Fragmente mit mehr als 4 Leucinen werden in Membran eingefügt. d, Mean probability of insertion (p) for H-segments with n = 0–7 Leu residues. For n = 0, 1, 5–7, only single H-segments with the following compositions were used (flanked by GGPG…GPGG in all cases): (A)19, (A)9L(A)9, (A)4LALAALAALAL(A)4, (A)4(LA)5L(A)4, ALAALALAALAALALAALA. Hessa et al., Nature 433, 377 (2005)
 for H-segments with n = 0–7 Leu residues. For n = 0, 1, 5–7, only single H-segments with the following compositions were used (flanked by GGPG…GPGG in all cases): (A)19, (A)9L(A)9, (A)4LALAALAALAL(A)4, (A)4(LA)5L(A)4, ALAALALAALAALALAALA. Hessa et al., Nature 433, 377 (2005)")
38
Vergleich von biologischen und Peptid G-Skalen
a, Gapp aa scale derived from H-segments with the indicated amino acid placed in the middle of the 19-residue hydrophobic stretch. c, Correlation between the Gapp aa scale and the Wimley–White water/octanol free energy scale. Fazit: Insertion in Membran hängt nur von Hydrophobizität der Aminosäuresequenz ab! Bioinformatiker sollten die Eigenschaften der Aminosäuren kennen Hessa et al., Nature 433, 377 (2005)
")
39
Helikale Räder Helikale Räder dienen zur Darstellung von Helices.
Man kann so leicht erkennen, welche Seite der Helix dem Solvens zugewandt ist und welche ins Proteininnere zeigt.

40
Analyse einer unbekannten Sequenz
Experimentelle Daten vorhanden? Input: neue Proteinsequenz Multiples Sequenzalignment Suche in Sequenzdatenbanken nach identischer Sequenz bzw. ähnlichen Sequenzen Erkenne Domänen Gibt es ähnliche Sequenz mit bekannter 3D-Struktur? Vorhersage der Sekundärstruktur Zuordnung eines Protein-Folds Nein Analyse dieses Folds, Nachbarn? Ja Ja Fold erkannt? Alignment der Sekundärstrukturen. Nein Modellierung der Proteinstruktur durch Homologiemodellierung Alignment der Sequenz mit einer Struktur. Vorhersage der Tertiärstruktur 3D-Proteinstruktur Kann man Funktion transferieren? Nach Rob Russell, gtsp/flowchart2.html

41
Netzwerke 1

42
Virtual Cell

43
Virtual Cell Left: overall mechanism of Ran-mediated nucleocytoplasmic transport. The image Right: membrane transport components within the Virtual Cell software. GTP-bound Ran shuttles between the nuclear and cytoplasmic compartments and is predominately nuclear at steady-state. The RanGTP nuclear membrane gradient is essential and required for RanGTP-dependent assembly and dissociation of transport complexes within the nucleus.

44
metabolische Netzwerke Formulierung von Biochemie mit Linearer Algebra

45
Software In den Tutorials vorgestellte Software:
0 Datenbankennavigation SRS I Sequenzanalyse: BLAST, PSI-BLAST, CLUSTALW II Proteinstruktur: VMD, Swissmodel III Zellsimulationen: Virtual Cell, FluxAnalyzer, Cytoscape Datenbanken: Sequenzdatenbanken Proteinstrukturbanken Metabolische Datenbanken

Ähnliche Präsentationen





















 Matrixkettenprodukt Prof. Dr. Th. Ottmann.>")

