Präsentation herunterladen
Die Präsentation wird geladen. Bitte warten

1
Softwarewerkzeuge der Bioinformatik
Inhalt dieser Veranstaltung: Softwarewerkzeuge für I Sequenzanalyse II Analyse von Proteinstruktur und Ligandenbindung III Zell- bzw. Netzwerksimulationen

2
„Lernziele“ Lerne aktuelle und bewährte Programme und Datenbanken der Bioinformatik kennen und erfolgreich einzusetzen um Tools kennenzulernen, mit denen man bioinformatische Fragen bearbeiten kann zu wissen, was auf dem Markt ist („das Rad nicht zweimal erfinden“) ein Gefühl dafür zu bekommen, wie erfolgreiche Softwareprodukte aussehen (sollen) 3 Mini-Forschungsprojekte zu bearbeiten Wir werden in der Vorlesung anhand von „Case-studies“ typische Fragestellungen in Pharma- oder Biotech-Unternehmen behandeln. Wie stellen Sie sich den Arbeitsalltag als Bioinformatiker in einer Pharma-Firma vor?
 ein Gefühl dafür zu bekommen, wie erfolgreiche Softwareprodukte. aussehen (sollen) 3 Mini-Forschungsprojekte zu bearbeiten. Wir werden in der Vorlesung anhand von „Case-studies typische Fragestellungen in Pharma- oder Biotech-Unternehmen behandeln. Wie stellen Sie sich den Arbeitsalltag als Bioinformatiker in einer Pharma-Firma vor")
3
Organisatorisches Jede Woche zweistündige Vorlesung Freitag 9-11, Hörsaal 1, Geb. 45 Dozent: Prof. Helms Übungen „hands-on“ im CIP-Pool Bioinformatik Raum R 104 im Geb. 45 Freitag Uhr. Betreuer der Übungen Sequenz-Analyse Sam Ansari Proteinstruktur Dr. Michael Hutter Zellsimulationen Dr. Tihamer Geyer

4
Welche Bioinformatik-Software gibt es?

11
Ein paar Produkte ...

12
Kommerzielle Software-Pakete sind bereits sehr mächtig
Kommerzielle Software ist sehr teuer, aber sehr mächtig, da integriert. Es ist fraglich, ob man in einer universitären Umgebung (mit kostenloser Software) bei Anwendungen im Bereich Drug Development mit Firmen konkurrieren kann, die solch mächtige Tools einsetzen.
 bei Anwendungen im Bereich Drug. Development mit Firmen konkurrieren. kann, die solch mächtige Tools einsetzen.")
13
Einsatz von Bioinformatik in der Produkt-Pipeline

14
Organisatorisches Jeder Teilnehmer an den Übungen benötigt einen Rechneraccount für den CIP-Pool. Diese Accounts werden von der Rechnerbetriebsgruppe des FB Informatik eingerichtet. Haben Sie bereits einen Account auf Uni-Rechnern? Dann muss dieser lediglich für den CIP-Pool freigeschaltet werden. Zugang zum CIP-Pool: Für Bioinformatik-Studenten 24/7, für alle anderen während der Übungsstunden. Bitte melden Sie sich nach dieser Stunde im Sekretariat des Zentrums für Bioinformatik bei Frau Alexandra Klasen an. Der Beginn der Übungen ist diese Woche.

15
Organisatorisches: Scheinvergabe
Die Vorlesung zählt 2V + 2P = 9 Leistungspunkte. Sie kann nach der neuen Prüfungsordnung für den Bachelor-Studiengang in der Vertiefung „Bioinformatics“ eingebracht werden. Die Scheine werden benotet. 50% der Benotung ergibt sich aus der mittleren Benotung von drei praktischen Aufgaben, die während des Semesters von jedem Studenten einzeln zu bearbeiten sind. Die Aufgaben werden etwa alle 4 Wochen ausgegeben und sind innerhalb von 2 Wochen zu bearbeiten und durch ein mindestens 5-seitiges Protokoll zu dokumentieren. Jeder Student muss mindestens zwei der drei praktischen Aufgaben mit einer Note von 4 und besser bestehen. Am Ende des Semesters wird eine 2-stündige Klausur über die Inhalte der Vorlesung und der Übungen geschrieben. Die Klausurnote geht ebenfalls mit 50% in die Scheinnote mit ein. Die Klausur muss mit einer Note von 4 und besser bestanden werden.

16
Übersicht über Vorlesungsinhalt
I Sequenz Einführung Paarweises Sequenzalignment Multiples Sequenzalignment Datenbanken Genomweite Sequenzanalyse II Struktur Proteinstruktur Proteinstrukturvorhersage Liganden-Docking Protein-Protein-Docking III Zellsimulationen E-Cell Virtual Cell Microarrays Protein-Netzwerke

17
Was fange ich mit diesen Daten an?
Sequenz des menschlichen Genoms wurde 2001 entschlüsselt.

18
1 2 Analyse einer unbekannten Sequenz
Input: neue Proteinsequenz Experimentelle Daten vorhanden? Multiples Sequenzalignment Suche in Sequenzdatenbanken nach identischer Sequenz bzw. ähnlichen Sequenzen Erkenne Domänen Gibt es ähnliche Sequenz mit bekannter 3D-Struktur? Nein Vorhersage der Sekundärstruktur Zuordnung eines Protein-Folds Analyse dieses Folds, Nachbarn? Ja Ja Fold erkannt? Alignment der Sekundärstrukturen. Nein Modellierung der Proteinstruktur durch Homologiemodellierung Alignment der Sequenz mit einer Target-Struktur Ab inito Vorhersage der Tertiärstruktur 3D-Proteinstruktur Nach Rob Russell, gtsp/flowchart2.html Kann man Funktion zuordnen?

19
Sequenzanalyse 1

20
Ziele (0) Identifiziere alle menschlichen Proteine (ORFs) und ihre Funktion Sind dies alle Proteine? Nein: post-translationelle Modifikationen möglich wie Methylierung, Phosphorylierung, Glykosilierung … Identifiziere Gen-Netzwerke. Welche Proteine wechselwirken miteinander? (2) Identifiziere Module: abgeschlossene Einheiten (3) Identifiziere Sequenz-Abschnitte, in denen Mutationen für Krankheiten codieren
 Identifiziere Module: abgeschlossene Einheiten. (3) Identifiziere Sequenz-Abschnitte, in denen Mutationen für Krankheiten codieren.")
21
Sequenzen sind verwandt
Evolution findet auf vielen verschiedenen Ebenen statt : Mutationen einzelner Aminosäuren, Domänen-Shuffling, Genduplikation, Genom-Rearrangement verwandte Moleküle besitzen in verschiedenen Organismen ähnliche Funktionen (“Homologe”) Phylogenetischer Baum für ribosomale RNA: Drei Bereiche des Lebens
 Phylogenetischer Baum für ribosomale RNA: Drei Bereiche des Lebens.")
22
Sequenzen sind verwandt, II
Phylogenetischer Baum für Globin-Proteine des Menschen

23
gewinne transferierbare Information aus Sequenzvergleich
Bestimme evolutionäre Beziehungen Vorhersage von Proteinfunktion und -struktur (Datenbanksuche). Protein 1: bindet Sauerstoff Sequenzähnlichkeit Protein 2: bindet Sauerstoff ?
. Protein 1: bindet Sauerstoff. Sequenzähnlichkeit. Protein 2: bindet Sauerstoff")
24
gap = Insertion oder Deletion
Sequenzalignment Der Zweck eines Sequenzalignments ist, all die Residuen einer beliebigen Anzahl von Sequenzen untereinander anzuordnen, die von der gleichen Residuenposition in einem Gen- oder Protein-Vorfahren abstammen. gap = Insertion oder Deletion Wat is het belangrijkste residue voor alignen? Cys, want meest geconserveerd A multiple sequence alignment is a 2D table, in which the rows represent individual sequences, and the columns the residue positions. Sequences are laid onto this grid in such a manner that (a) the relative positioning of residues within any one sequence is preserved, and (b) similar residues in all the sequences are brought into vertical register.
 the relative positioning of residues within any one sequence is preserved, and (b) similar residues in all the sequences are brought into vertical register.")
25
Needleman-Wunsch Algorithmus
allgemeiner Algorithmus für Sequenzvergleiche maximiert einen Ähnlichkeitsscore bester Match = grösste Anzahl an Residuen einer Sequenz, die zu denen einer anderen Sequenz passen, wobei Deletionen erlaubt sind. Der Algorithmus findet durch dynamische Programmierung das bestmögliche GLOBALE Alignment zweier beliebiger Sequenzen NW beinhaltet eine iterative Matrizendarstellung alle möglichen Residuenpaare (Basen oder Aminosäuren) – je eine von jeder Sequenz – werden in einem zwei-dimensionalen Gitter dargestellt. alle möglichen Alignments werden durch Pfade durch dieses Gitter dargestellt. Der Algorithmus hat 3 Schritte: 1 Initialisierung 2 Auffüllen 3 Trace-back What database sequences are most similar to (or contain the most similar regions to) my previously uncharacterised sequence? Searching a data base needs to be fast and sensitive but the two objectives counteract each other and has a high sensitivity for detecting distant sequence relationships between a query sequence and a database. Input=seq Output= list of seq that match query sequence
 – je eine von jeder Sequenz – werden in einem zwei-dimensionalen Gitter dargestellt. alle möglichen Alignments werden durch Pfade durch dieses Gitter dargestellt. Der Algorithmus hat 3 Schritte: 1 Initialisierung 2 Auffüllen 3 Trace-back. What database sequences are most similar to (or contain the most similar regions to) my previously uncharacterised sequence Searching a data base needs to be fast and sensitive but the two objectives. counteract each other. and has a high sensitivity for detecting distant sequence relationships between a query sequence and a database. Input=seq. Output= list of seq that match query sequence.")
26
Needleman-Wunsch Algorithm: Initialisierung
Aufgabe: aligniere die Wörter “COELACANTH” und “PELICAN” der Länge m =10 und n =7. Konstruiere (m+1) (n+1) Matrix. Ordne den Elementen der ersten Zeile und Reihe die Werte – m gap und – n gap zu. Die Pointer dieser Felder zeigen zurück zum Ursprung. C O E L A N T H -1 -2 -3 -4 -5 -6 -7 -8 -9 -10 P I What database sequences are most similar to (or contain the most similar regions to) my previously uncharacterised sequence? Searching a data base needs to be fast and sensitive but the two objectives counteract each other and has a high sensitivity for detecting distant sequence relationships between a query sequence and a database. Input=seq Output= list of seq that match query sequence
 (n+1) Matrix. Ordne den Elementen der ersten Zeile und Reihe die Werte – m gap und – n gap zu. Die Pointer dieser Felder zeigen zurück zum Ursprung. C. O. E. L. A. N. T. H P. I. What database sequences are most similar to (or contain the most similar regions to) my previously uncharacterised sequence Searching a data base needs to be fast and sensitive but the two objectives. counteract each other. and has a high sensitivity for detecting distant sequence relationships between a query sequence and a database. Input=seq. Output= list of seq that match query sequence.")
27
Needleman-Wunsch Algorithm: Auffüllen
Fülle alle Matrizenfelder mit Werten und Zeigern gemäss von simplen Operationen, die die Werte der diagonalen, vertikal, und horizontalen Nachbarzellen einschliessen. Berechne match score: Wert der Diagonalzelle links oben + Wert des Alignments (+1 oder -1) horizontal gap score: Wert der linken Zelle + gap score (-1) vertical gap score: Wert der oberen Zelle + gap score (-1) ordne der Zelle das Maximum dieser 3 Werte zu. Der Pointer zeigt in Richtung des maximalen Scores. max(-1, -2, -2) = -1 max(-2, -2, -3) = -2 (Pointer soll bei gleichen Werte immer in eine bestimmte Richtung zeigen, z.B. entlang der Diagonalen. C O E L A N T H -1 -2 -3 -4 -5 -6 -7 -8 -9 -10 P What database sequences are most similar to (or contain the most similar regions to) my previously uncharacterised sequence? Searching a data base needs to be fast and sensitive but the two objectives counteract each other and has a high sensitivity for detecting distant sequence relationships between a query sequence and a database. Input=seq Output= list of seq that match query sequence
 horizontal gap score: Wert der linken Zelle + gap score (-1) vertical gap score: Wert der oberen Zelle + gap score (-1) ordne der Zelle das Maximum dieser 3 Werte zu. Der Pointer zeigt in Richtung des maximalen Scores. max(-1, -2, -2) = -1. max(-2, -2, -3) = -2. (Pointer soll bei gleichen Werte immer in eine bestimmte Richtung zeigen, z.B. entlang der Diagonalen. C. O. E. L. A. N. T. H P. What database sequences are most similar to (or contain the most similar regions to) my previously uncharacterised sequence Searching a data base needs to be fast and sensitive but the two objectives. counteract each other. and has a high sensitivity for detecting distant sequence relationships between a query sequence and a database. Input=seq. Output= list of seq that match query sequence.")
28
Needleman-Wunsch Algorithmus: Trace-back
Trace-back ergibt das Alignment aus der Matrix. Starte in Ecke rechts unten und folge den Pfeilen bis in die Ecke links oben. COELACANTH -PELICAN-- C O E L A N T H -1 -2 -3 -4 -5 -6 -7 -8 -9 -10 P I 1 2 What database sequences are most similar to (or contain the most similar regions to) my previously uncharacterised sequence? Searching a data base needs to be fast and sensitive but the two objectives counteract each other and has a high sensitivity for detecting distant sequence relationships between a query sequence and a database. Input=seq Output= list of seq that match query sequence
 my previously uncharacterised sequence Searching a data base needs to be fast and sensitive but the two objectives. counteract each other. and has a high sensitivity for detecting distant sequence relationships between a query sequence and a database. Input=seq. Output= list of seq that match query sequence.")
29
Smith-Waterman-Algorithmus
Smith-Waterman ist ein lokaler Alignment-Algorithmus. SW ist eine sehr einfache Modifikation von Needleman-Wunsch. Lediglich 3 Änderungen: die Matrixränder werden auf 0 statt auf ansteigende Gap-Penalties gesetzt. der maximale Wert sinkt nie unter 0. Pointer werden nur für Werte grösser als 0 eingezeichnet. Trace-back beginnt am grösseten Wert der Matrix und endet bei dem Wert 0. ELACAN ELICAN C O E L A N T H P 1 2 I 3 4 What database sequences are most similar to (or contain the most similar regions to) my previously uncharacterised sequence? Searching a data base needs to be fast and sensitive but the two objectives counteract each other and has a high sensitivity for detecting distant sequence relationships between a query sequence and a database. Input=seq Output= list of seq that match query sequence
 my previously uncharacterised sequence Searching a data base needs to be fast and sensitive but the two objectives. counteract each other. and has a high sensitivity for detecting distant sequence relationships between a query sequence and a database. Input=seq. Output= list of seq that match query sequence.")
30
Sequenzvergleiche: PAM250 Matrix
Für Sequenzvergleiche werden Scoring-Matrizen für den Austausch von Aminosäuren verwendet. 1) Notice 1 lettercode for the amino acids on both axes are the 20 aa note blocks of similar amino acids 2) Symmetric, only one half shown 3) Diagonal: * For example: high score for matching Tryptophans and “low” score for matching Alanines. * Cysteine * Leu abundant 4) Off-diagonal Groups of similar amino acids K -> F -5 A score above zero assigned to two amino acids indicates that these two .. Each other more often than expected by chance alone. Ie they are functionall.. Exchangable A negative score indicates that the two amino acids are rarely .. Interchangeable. Eg. A basic amino acids for an acidic one or one with an … side chain for one with aliphatic side chain.
 Notice 1 lettercode for the amino acids on both axes are the 20 aa note blocks of similar amino acids 2) Symmetric, only one half shown 3) Diagonal: * For example: high score for matching Tryptophans and low score for matching Alanines. * Cysteine. * Leu abundant. 4) Off-diagonal Groups of similar amino acids K -> F -5. A score above zero assigned to two amino acids indicates that these two .. Each other more often than expected by chance alone. Ie they are functionall.. Exchangable. A negative score indicates that the two amino acids are rarely .. Interchangeable. Eg. A basic amino acids for an acidic one or one with an … side chain for one with aliphatic side chain.")
31
Proteinstruktur Sequenz
Was hat nun Sequenz-Konservierung mit Proteinstrukturen zu tun? sehr viel! Die Twilight zone kennzeichnet das Mass an Sequenzidentität, bis zu der zwei Proteinstrukturen mit hoher Wkt. die gleiche Struktur besitzen. Richtlinien von Doolittle: Sequenzen mit > 150 Residuen und 25% Sequenzidentität sind wahrscheinlich verwandt mit 15-20% Sequenzidentität können sie verwandt sein bei <15% Sequenzidentität ist es schwierig zu sagen ob sie verwandt sind oder nicht ohne weitere strukturelle oder funktionelle Hinweise TWILIGHT ZONE 1

32
Wechselwirkung mit Liganden
Proteinstruktur, Wechselwirkung mit Liganden 1

33
Einleitung: Aminosäuren
Aminosäuren sind die Bausteine von Proteinen: Aminogruppe Carboxylsäure 1

34
Buchstaben-Code der Aminosäuren
Ein- und Drei-Buchstaben-Codes der Aminosäuren G Glycin Gly P Prolin Pro A Alanin Ala V Valin Val L Leucin Leu I Isoleucin Ile M Methionin Met C Cystein Cys F Phenylalanin Phe Y Tyrosin Tyr W Tryptophan Trp H Histidin His K Lysin Lys R Arginin Arg Q Glutamin Gln N Asparagin Asn E Glutaminsäure Glu D Asparaginsäure Asp S Serin Ser T Threonin Thr Zusätzliche Codes B Asn/Asp Z Gln/Glu X Irgendeine Aminosäure

35
Einleitung: Peptidbindung
In Peptiden und Proteinen sind die Aminosäuren miteinander als lange Ketten verknüpft. Ein Paar ist jeweils über eine „Peptidbindung“ verknüpft. Die Aminosäuresequenz eines Proteins bestimmt seinen „genetischen code“. Die Kenntnis der Sequenz eines Proteins allein verrät noch nicht viel über seine Funktion. Entscheidend ist seine drei-dimensionale Struktur. 1

36
Grundlegende Definitionen
Primärstruktur Die lineare Sequenz der Aminosäuren eines Proteins Sekundärstruktur Regionen lokaler Regelmässigkeit Z.B. -Helices, -Stränge, -Faltblätter & -Schleifen

37
Definition: Super-Sekundärstruktur
Die Anordnung (packing) von Sekundärstrukturelemente zu stabilen Einheiten wie b-barrels, bab Einheiten, Greek keys, usw.
 von Sekundärstrukturelemente zu stabilen Einheiten. wie b-barrels, bab Einheiten, Greek keys, usw.")
38
Definition: Tertiärstruktur
Die gesamte Faltung einer Kette, die sich aus der Packung der Sekundärstrukturelemente ergibt. Grün Fluoreszierendes Protein. Seine zylindrische Architektur wird durch 11 -Stränge gebildet. (1emb.pdb Brejc et al. 1997)
")
39
Einleitung: Proteinstruktur
cAMP-abhängige Proteinkinase Ca2+ Pumpe (katalytische Untereinheit) (TM Protein) 1
 (TM Protein) 1.")
40
Definition: Quartäre Struktur
Die Anordnung mehrerer Ketten eines Proteins, das mehrere Untereinheiten besitzt. Beispiel Hämoglobin

41
Bedeutung von Sequenzanalyse
>900,000 Sequenzen in öffentlichen Datenbanken zugänglich Millionen mehr in proprietären dbs Anstieg wird mit Sequenzierung von weiteren Genomen weitergehen Was tun? In den Sequenzen steckt eine grosse Menge an strukturellen, funktionellen und evolutionären Informationen Sie sind eine sehr wichtige Datenquelle Im Gegensatz dazu gibt es nur etwa 2000 unabhängige Proteinstrukturen

42
Sequenz-Struktur Missverhältnis
800 700 600 500 400 300 200 100 Anzahl an nicht-redundanten Sequenzen ( ) Entsprechende Zunahme der Zahl an Proteinstrukturen ( ).
 Entsprechende Zunahme der Zahl an Proteinstrukturen ( ).")
43
Der “holy grail” der strukturellen Bioinformatik

44
Eigenschaften der Aminosäuren
Aminosäuren unterscheiden sich in ihren physikochemischen Eigenschaften.

45
Einleitung: hydrophobe Aminosäuren
Proteine sind aus 20 verschiedenen natürlichen Aminosäuren aufgebaut 5 sind hydrophob. Sie sind vor allem Im Proteininneren. 1

46
Einleitung: aromatische Aminosäuren
Es gibt drei voluminöse aromatische Aminosäuren. Tyrosin und Tryptophan liegen bei Membranproteinen vor allem in der Interface-region. 1

47
Einleitung: Aminosäuren
Es gibt 2 Schwefel enthaltende Aminosäuren und das ungewöhnliche Prolin. Cysteine können Disulfidbrücken bilden. Prolin ist ein “Helixbrecher”. 1

48
Einleitung: Aminosäuren
Es gibt zwei Aminosäuren mit terminalen polaren Hydroxlgruppen: 1

49
Einleitung: Aminosäuren
Es gibt 3 positiv geladene Aminosäuren. Sie liegen vor allem auf der Proteinoberflächen und in aktiven Zentren. Thermophile Organismen besitzen besonders viele Ionenpaare auf den Protein-oberflächen. 1

50
Einleitung: Aminosäuren
Es gibt 2 negativ geladene Aminosäuren und ihre zwei neutralen Analoga. Asp und Glu haben pKa Werte von 2.8. Das heisst, erst unterhalb von pH=2.8 werden ihre Carboxylgruppe protoniert. 1

51
Transmembrandomänen: Hydrophobizitätsskalen
Stephen White group, UC Irvine

52
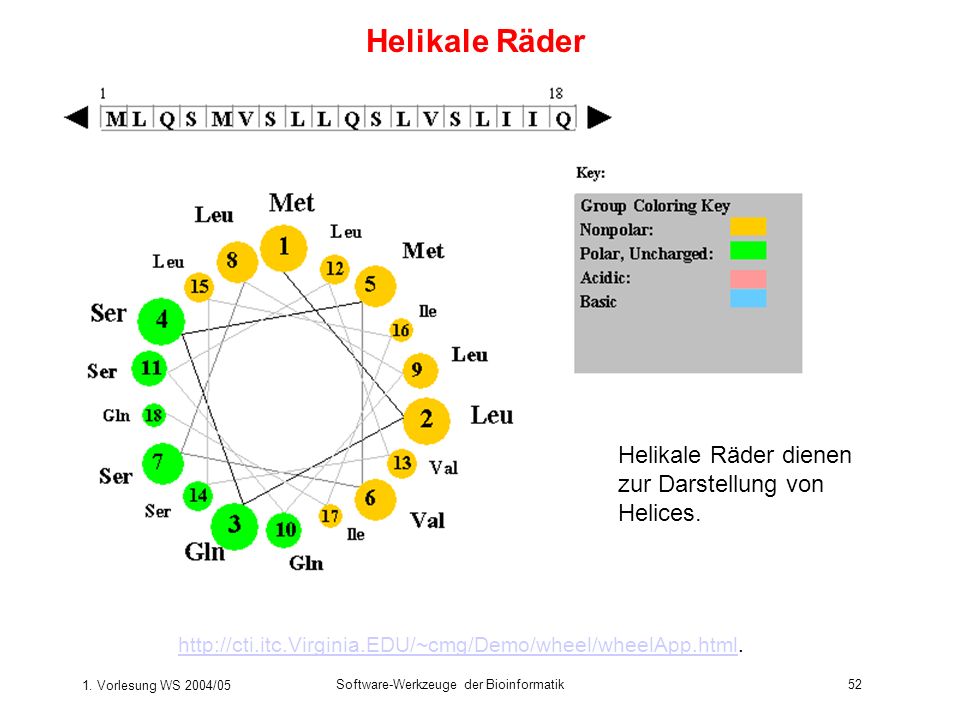
Helikale Räder Helikale Räder dienen zur Darstellung von Helices.
53
Analyse einer unbekannten Sequenz
Experimentelle Daten vorhanden? Input: neue Proteinsequenz Multiples Sequenzalignment Suche in Sequenzdatenbanken nach identischer Sequenz bzw. ähnlichen Sequenzen Erkenne Domänen Gibt es ähnliche Sequenz mit bekannter 3D-Struktur? Vorhersage der Sekundärstruktur Zuordnung eines Protein-Folds Nein Analyse dieses Folds, Nachbarn? Ja Ja Fold erkannt? Alignment der Sekundärstrukturen. Nein Modellierung der Proteinstruktur durch Homologiemodellierung Alignment der Sequenz mit einer Struktur. Vorhersage der Tertiärstruktur 3D-Proteinstruktur Kann man Funktion transferieren? Nach Rob Russell, gtsp/flowchart2.html

54
Proteinstruktur Sequenz
Konservierung von Residuen sind Indizien für den Verwandtschaftsgrad von Proteinen, für die Evolution und für die Verwandtschaft von Organismen Konservierung von Residuen im aktiven Zentrum Konservierung von Residuen, die die Architektur der Proteinstruktur stabilisieren Konservierung von Residuen, die während Faltung des Proteins wichtig sind Konservierung von Residuen an Bindungsschnittstellen für Liganden und andere Proteine 1

55
Netzwerke 1

56
metabolische Netzwerke Formulierung von Biochemie mit Linearer Algebra

57
Zellsimulationen Ziel: verstehe metabolische Abläufe in Zellen

58
E-cell Anwendungen bisher: - Energie-Metabolismus von E.coli - e-Rice
- Modell eines menschlichen Erythrozyten - Zirkadiane Rhythmen - e-Neuron - Signalübertragung in der bakteriellen Chemotaxis

59
Virtual Cell

60
Virtual Cell Left: overall mechanism of Ran-mediated nucleocytoplasmic transport. The image Right: membrane transport components within the Virtual Cell software. GTP-bound Ran shuttles between the nuclear and cytoplasmic compartments and is predominately nuclear at steady-state. The RanGTP nuclear membrane gradient is essential and required for RanGTP-dependent assembly and dissociation of transport complexes within the nucleus.

61
Virtual Cell Parameter …

62
Virtual Cell This set of images shows the spatiotemporal pattern of nuclear accumulation of fluorescently labeled Ran after microinjection into the cytosol in a confocal experiment (grayscale panels) and a Virtual Cell simulation (color scale panels).
 and a Virtual Cell simulation (color scale panels).")
63
Virtual Cell Calculated 3D distribution of 2 species in the pathway that are not directly visible by labeling. Injecting fluorescently labeled Ran allows you to experimentally visualize all the forms of Ran but not the individual bound and free states. Simulations help dissect what is happening to all the species.

64
Software In den Tutorials vorgestellte Software:
0 Datenbankennavigation SRS I Sequenzanalyse: (FASTA) BLAST, PSI-BLAST, CLUSTALW II Proteinstruktur: VMD Ligandenbindung: FlexX mit Andreas Kämper III Zellsimulationen: Virtual Cell Datenbanken: Sequenzdatenbanken Proteinstrukturbanken Metabolische Datenbanken
 BLAST, PSI-BLAST, CLUSTALW. II Proteinstruktur: VMD. Ligandenbindung: FlexX mit Andreas Kämper. III Zellsimulationen: Virtual Cell. Datenbanken: Sequenzdatenbanken. Proteinstrukturbanken. Metabolische Datenbanken.")
Ähnliche Präsentationen













>")










