Präsentation herunterladen
Die Präsentation wird geladen. Bitte warten

1
Morbus Parkinson-Frühdiagnose, Initialtherapie und Neuroprotektion
Wien, Heinz Reichmann, FRCP, FAAN Neurologische Klinik, Technische Universität Dresden

2
Wie lange dauert es, bis de-novo Patienten diagnostiziert werden?
Average time in months from onset of symptoms to initial presentation Datamonitor; 2007

3
Könnte eine frühere Diagnose zu einer anderen Therapie führen?
2 2: Möglich, wenn klinische Tets entwickelt werden, die erste Symptome früher identifiziert werden können, die heute noch als unspezifisch angesehen werden. 3 3:Zunehmend plausibel, die frühesten pathologischen Veränderungen zu entdecken (bevor sich Symptome entwickeln), was ermöglicht, neuroprotektive Agentien so früh wie möglich einzusetzen. 1: Diagnose und Behandlung beschleunigt durch frühere Vorstellung von Patienten mit characteristischem Phänotyp und Verbesserung der diagnostischen Fähigkeiten 1 Prä-diagnostische Phase Behandlung Pathologishe Progression Prä-symptomatisch Early and differential diagnosis of PD is key to its effective management, as a delayed or incorrect diagnosis can be a barrier to the provision of appropriate treatment and counselling.1-3 In addition, an early diagnosis could also offer the opportunity to administer disease-modifying therapy, and potentially address the rate of disease progression from an early stage.2 The development of subtle clinical correlates provides an opportunity to make an early diagnosis before the onset of characteristic extrapyramidal motor symptoms.3 At some point after the development of PD, the pathological process begins, but symptoms do not emerge until ~50% of the dopaminergic cells in the substantia nigra are lost, a process estimated to take ~5 years.3 At first, the symptoms are rather non-specific, with an estimated 4–6 year lag period until the diagnosis of PD is made on the basis of the characteristic symptoms that eventually develop.3 The overall rate of disease progression (slope of the graph) will differ between patients, and is also likely to alter over time within a single patient (making it non-linear).3 The diagnosis and subsequent treatment of patients can be expedited if patients present earlier with the characteristic phenotype of disease, and if our clinical diagnostic skills are improved (1).3 Alternatively, it may be possible to develop clinical tests that can positively identify the earlier symptoms that are currently regarded as non-specific (2).3 Finally, it seems increasingly plausible that it will be feasible to detect the earliest pathological changes even before symptoms develop, potentially allowing the use of neuroprotective agents as early as possible (3).3 1. Tolosa E, Compta Y, Gaig C. Parkinsonism Relat Disord 2007; 13: S2–S7. 2. Schapira AHV, Obeso J. Ann Neurol 2006; 59 (3): 559–564. 3. Michell AW, Lewis SJG, Foltynie T, Barker RA. Brain 2004; 127: 1693–1705 Zeit Pathologischer Beginn Nicht-spezifisch Spezifisch Diagnose Symptom- Beginn Michell, et al. Brain 2004; 127: 1693 3
, was ermöglicht, neuroprotektive Agentien so früh wie möglich einzusetzen. 1: Diagnose und Behandlung beschleunigt durch frühere Vorstellung von Patienten mit characteristischem Phänotyp und Verbesserung der diagnostischen Fähigkeiten. 1. Prä-diagnostische Phase. Behandlung. Pathologishe Progression. Prä-symptomatisch. Early and differential diagnosis of PD is key to its effective management, as a delayed or incorrect diagnosis can be a barrier to the provision of appropriate treatment and counselling.1-3 In addition, an early diagnosis could also offer the opportunity to administer disease-modifying therapy, and potentially address the rate of disease progression from an early stage.2 The development of subtle clinical correlates provides an opportunity to make an early diagnosis before the onset of characteristic extrapyramidal motor symptoms.3. At some point after the development of PD, the pathological process begins, but symptoms do not emerge until ~50% of the dopaminergic cells in the substantia nigra are lost, a process estimated to take ~5 years.3. At first, the symptoms are rather non-specific, with an estimated 4–6 year lag period until the diagnosis of PD is made on the basis of the characteristic symptoms that eventually develop.3 The overall rate of disease progression (slope of the graph) will differ between patients, and is also likely to alter over time within a single patient (making it non-linear).3. The diagnosis and subsequent treatment of patients can be expedited if patients present earlier with the characteristic phenotype of disease, and if our clinical diagnostic skills are improved (1).3 Alternatively, it may be possible to develop clinical tests that can positively identify the earlier symptoms that are currently regarded as non-specific (2).3 Finally, it seems increasingly plausible that it will be feasible to detect the earliest pathological changes even before symptoms develop, potentially allowing the use of neuroprotective agents as early as possible (3) Tolosa E, Compta Y, Gaig C. Parkinsonism Relat Disord 2007; 13: S2–S7. 2. Schapira AHV, Obeso J. Ann Neurol 2006; 59 (3): 559– Michell AW, Lewis SJG, Foltynie T, Barker RA. Brain 2004; 127: 1693–1705. Zeit. Pathologischer Beginn. Nicht-spezifisch. Spezifisch. Diagnose. Symptom- Beginn. Michell, et al. Brain 2004; 127:")
4
Genetisches Screening
Moore et al. (2005)
")
5
LRRK2-Punktmutation

6
Penetranz-Risiko bei LRRK2-PM
Figure 3: Age-specific risk of PD Risk is estimated with the Kaplan-Meier method for the whole sample and with the maximum-likelihood estimation (ML) for all patients with mutations in LRRK2 combined. Healy et al. (2008)
 for all patients with mutations in LRRK2 combined. Healy et al. (2008)")
7
Die Braak Stadien Schematic diagrams showing the gradual ascent of the pathologic process underlying IPD. b. During the presymptomatic stages 1 and 2, the IPD-related inclusion body pathology is confined to the medulla oblongata and olfactory bulb. c. In stages 3 and 4, the substantia nigra and other nuclear grays of the midbrain and basal forebrain become the focus of initially subtle and, then, severe changes. The illness most probably reaches its symptomatic phase in many individuals. d. In the final stages 5 and 6, the lesions encroach upon the cerebral cortex, so that IPD manifests itself in all of its aspects: somatomotor dysfunctions are supplemented by increasing deterioration of cortically controlled intellectual capabilities. Braak et al .

8
Riechstifte

9
Prävalenz der Hyposmie bei PD

10
Transcranial sonography in idiopathic olfactory dysfunction
Individual results of testing normal borderline pathological test not performed Conversion to IPD definitive borderline UPDRS II III TCS SPECT 123I-FP-CIT (DaTScan) UPDRS III Haehner et al. Mov Disord. 2007;22:
 UPDRS III. Haehner et al. Mov Disord. 2007;22:")
11
Präklinische Detektion
normosmic n = 40 hyposmic n = 283 baseline 2 Jahre n = 33 n = 279 n = 39 n = 361 asymptomatic relatives 50-75 Jahre 12 % accelerated loss of DAT binding 10 % parkinsonism 0 % accelerated loss of DAT binding 0 % parkinsonism n = 1 n = 3 Ponsen et al. Ann Neurol 2004;56:

12
Aetiopathogenese

13
a-Synuclein Einschlüsse im ENS
Presence of gastric a-synuclein inclusions could provide first link in susceptible neurons that extend from the enteric to the central nervous system individuals.

14
Das Dresdner Parkinson Modell

15
Methoden und Ergebnisse
Rotenon ist ein Zellgift, das den Komplex I der Atmungskette in den Mitochondrien inhibiert Administration von Rotenon intragastral über einen Magenschlauch im Mäusemodell Rotenon konnte dabei weder im Blut noch im Gehirn nachgewiesen werden, somit erzielten wir eine rein lokale Wirkung Weder in der Muskulatur noch im Gehirn der Tiere zeigte sich eine Komplex I-Hemmung A-synuclein Aggregation konnte im ENS von „behandelten“ Tieren nachgewiesen werden Danach konnte a-Synuclein auch im Nucleus intermediolateralis im RM und im dorsalen motorischen Kern des Nervus vagus detektiert werden Nach 3 Monaten kam es zu typischen a-Synuclein-Ablagerungen in der SN und zu einer Abnahme von 15% an TH-pos Neuronen in der SN. Zu diesem Zeitpunkt waren die Tiere auf dem Laufrad signifikant verlangsamt

16
Befund im Nc. vagus Treated (DMV) A-synuclein Con 1,5 mo 3mo Control
Figure 3. Intragastrically administered rotenone induces alpha-synuclein accumulation, oxidative stress and inflammation in the dorsal motor nucleus vagus. (scale bars 20 um). A, B, double-immunofluorescence staining against alpha-synuclein and ChAT on DMV sections from 1.5 months control (A) and 1.5 months treated (B) mice. Arrows in B, increased intracellular alpha-synuclein in DMV neurons already after 1.5 months. Arrowheads in B, autofluorescent punctate inclusion pattern inside ChAT+ neurons. C, DMV sections stained with ChAT and DAPI were sequentially excited with 488 and 561 laser wavelengths. Arrows in C, large intracellular auto-fluorescent inclusions inside ChAT+ neurons of the DMV (arrows). D, E, F, Light microscopy images of alpha-synuclein staining from 1.5 months control (D), 1.5 months (E) and 3 months (F) treated mice. Arrows in E and F, increased staining intensity inside DMV neuronal soma in treated mice. Arrowheads in F, increased alpha-synuclein staining inside neuronal processes G, H, average-projection of triple-immunofluorescence staining against ChAT, GFAP, MHC II (clone M5/ ) and DAPI on sections from control (G) and treated (H) mice after 3 month treatment. Arrow in H, activated microglial cell in the DMV. Control Treated (DMV) A-synuclein Con 1,5 mo 3mo Pan-Montojo et al. (2010)
. A, B, double-immunofluorescence staining against alpha-synuclein and ChAT on DMV sections from 1.5 months control (A) and 1.5 months treated (B) mice. Arrows in B, increased intracellular alpha-synuclein in DMV neurons already after 1.5 months. Arrowheads in B, autofluorescent punctate inclusion pattern inside ChAT+ neurons. C, DMV sections stained with ChAT and DAPI were sequentially excited with 488 and 561 laser wavelengths. Arrows in C, large intracellular auto-fluorescent inclusions inside ChAT+ neurons of the DMV (arrows). D, E, F, Light microscopy images of alpha-synuclein staining from 1.5 months control (D), 1.5 months (E) and 3 months (F) treated mice. Arrows in E and F, increased staining intensity inside DMV neuronal soma in treated mice. Arrowheads in F, increased alpha-synuclein staining inside neuronal processes G, H, average-projection of triple-immunofluorescence staining against ChAT, GFAP, MHC II (clone M5/ ) and DAPI on sections from control (G) and treated (H) mice after 3 month treatment. Arrow in H, activated microglial cell in the DMV. Control. Treated (DMV) A-synuclein. Con. 1,5 mo. 3mo. Pan-Montojo et al. (2010)")
17
Substantia nigra pars compacta
Con Pan-Montojo et al. (2010) Figure 4. Alpha-synuclein accumulation and neuronal loss in the SNc after 3 but not 1.5 months intragastrical rotenone treatment. (A–C, scale bars 20 um; E–F, scale bars 200 um). A, B, C, immunostaining against TH, alpha-synuclein and DAPI on SNc sections from 1.5 months control (A) and 3 months (B–C) treated mice. Arrow in B, alpha-synuclein small inclusions inside TH+ neurons. Arrow in C, large alpha-synuclein inclusion (|>8.14 um) inside a dopamineric neuron in the SN. D, stereological quantification (n = 3) of TH+ neurons in the SN from control and treated mice. Asterisk, P<0.05. Number of neurons was determined based on the optical fractionator principle using StereoInvestigator software (MicroBrightField Inc., Williston, USA). Each column represents total number of TH+ neurons in the SN in 1.5 and 3 months control and treated mice. Graph shows mean +/-s.e.m. E, F, TH immunostaining on striatum in 1.5 months control (E) and 3 months treated (F) mice. Th-stain in control € and treated mice (F)
 Figure 4. Alpha-synuclein accumulation and neuronal loss in the SNc after 3 but not 1.5 months intragastrical rotenone treatment. (A–C, scale bars 20 um; E–F, scale bars 200 um). A, B, C, immunostaining against TH, alpha-synuclein and DAPI on SNc sections from 1.5 months control (A) and 3 months (B–C) treated mice. Arrow in B, alpha-synuclein small inclusions inside TH+ neurons. Arrow in C, large alpha-synuclein inclusion (|>8.14 um) inside a dopamineric neuron in the SN. D, stereological quantification (n = 3) of TH+ neurons in the SN from control and treated mice. Asterisk, P<0.05. Number of neurons was determined based on the optical fractionator principle using StereoInvestigator software (MicroBrightField Inc., Williston, USA). Each column represents total number of TH+ neurons in the SN in 1.5 and 3 months control and treated mice. Graph shows mean +/-s.e.m. E, F, TH immunostaining on striatum in 1.5 months control (E) and 3 months treated (F) mice. Th-stain in control € and treated mice (F)")
18
Weitere Evidenz Hemivagotomy and partial sympathectomy delay Parkinson’s disease progression in mice Francisco Pan-Montojo1,2, 5, Mathias Schwarz1, Clemens Winkler1, Mike Arnhold2, Gregory O’Sullivan4, Arun Pal4, Margarita Rodrigo-Angulo5, Gabriele Gille2, Richard H.W. Funk1,3, and Heinz Reichmann2,3 1Institute for Anatomy, TU-Dresden, Fetscherstr. 74, 01307, Dresden 2Department of Neurology, University Hospital Carl-Gustav Carus, Fetscherstr. 74, 01307, Dresden, Germany 3Center for Regenerative Therapies Dresden, Tatzberg 47/49, 01307, Dresden, Germany 4Max-Planck Institute for Cell Biology and Genetics, Pfotenhauerstr. 108, 01307, Dresden, Germany 5Departamento de Anatomía, Histología y Neurociencia, Facultad de Medicina, Universidad Autónoma de Madrid, Arzobispo Morcillo 4, Madrid, Spain Abstract Pathological studies on Parkinson’s disease (PD) patients suggest that PD pathology starts at the olfactory bulb (OB) and the enteric nervous system (ENS) progressing into the central nervous system (CNS). In our previous study, we showed that the local effect of rotenone on the ENS reproduces this pathological progression in mice affecting only synaptically connected structures, suggesting transsynaptic and retrograde axonal transport as underlying mechanisms of this progression. Here, we tested this hypothesis by performing a hemivagotomy or a partial sympathectomy prior to rotenone oral treatment on mice and using primary enteric and sympathetic neuron co-cultures. For the first time, our results show that the appearance of motor dysfunctions is delayed in hemi-vagotomized and sympathectomized treated mice when compared to non-operated treated mice. Moreover, we only observed accumulation of alpha-synuclein in those structures still connected to the ENS. Interestingly, enteric neurons secrete alpha-synuclein only upon exposure to rotenone and secreted alpha-synuclein can be up-taken by non-neuronal cells or presynaptic sympathetic neurons. Altogether, these results suggest that pesticide-dependent alterations in the ENS can induce idiopathic PD pathology and trigger its progression. Moreover, it seems that this progression is based on the transsynaptic and retrograde axonal transport of alpha-synuclein, playing here the role of a prionic protein.
 patients suggest that PD pathology starts at the olfactory bulb (OB) and the enteric nervous system (ENS) progressing into the central nervous system (CNS). In our previous study, we showed that the local effect of rotenone on the ENS reproduces this pathological progression in mice affecting only synaptically connected structures, suggesting transsynaptic and retrograde axonal transport as underlying mechanisms of this progression. Here, we tested this hypothesis by performing a hemivagotomy or a partial sympathectomy prior to rotenone oral treatment on mice and using primary enteric and sympathetic neuron co-cultures. For the first time, our results show that the appearance of motor dysfunctions is delayed in hemi-vagotomized and sympathectomized treated mice when compared to non-operated treated mice. Moreover, we only observed accumulation of alpha-synuclein in those structures still connected to the ENS. Interestingly, enteric neurons secrete alpha-synuclein only upon exposure to rotenone and secreted alpha-synuclein can be up-taken by non-neuronal cells or presynaptic sympathetic neurons. Altogether, these results suggest that pesticide-dependent alterations in the ENS can induce idiopathic PD pathology and trigger its progression. Moreover, it seems that this progression is based on the transsynaptic and retrograde axonal transport of alpha-synuclein, playing here the role of a prionic protein.")
19
Aetiologie des Idiopathischen
Parkinson-Syndroms

20
REM-Schlaf-Verhaltensstörung (RBD)
Traum-assoziierte Bewegungen im Rahmen von RBD (fehlende Atonie)
")
21
Follow-up von Patienten mit idiopathischer RBD

22
Welcher Patient sollte behandelt werden?
Watch maker Bricklayer

23
Frühphase – eine kritische Zeit für die Krankheitsprogression
Klinische und Imaging Studien deuten darauf hin, dass die Frühphase nach der Diagnose entscheidend für die Progression ist Study Drug UPDRS loss/year in placebo group DATATOP Selegiline 14 ROADS Lazabemide 8.1 QE2 CoQ10 9 TEMPO Rasagiline 7.8 ELLDOPA L-DOPA 10.6 TCH346 7.6* It is becoming increasingly apparent that the early stage of PD is critical in terms of the rate of disease progression providing an opportunity for disease-modifying interventions. Imaging and pathology data have indicated that the presymptomatic phase of PD lasts ~4.5–6 years, with an exponential rate of dopaminergic cell loss in the preclinical and early clinical phases of the disease.1-4 Furthermore, the rate of clinical deterioration is rapid in early PD, with an annual decline of ~8–14 UPDRS points for patients with early PD.5-10 1. Schapira AHV, Obeso J. Ann Neurol 2006; 59 (3): 559–565. 2. Morrish PK, Sawle GV, Brooks DJ. Brain 1996; 119: 585–591. 3. Fearnley JM, Lees AJ. Brain 1991; 114: 2283–2301. 4. Hilker R, Schweitzer K, Coburger S, et al. Arch Neurol 2005; 62: 378 –382. 5. Parkinson Study Group. N Engl J Med 1993; 328: 176–183. 6. Parkinson Study Group. Ann Neurol 1996; 40: 99–107. 7. Shults et al. Arch Neurol 2002; 59: 1541–1550. 8. Parkinson Study Group. Arch Neurol 2002; 59: 1937–1943. 9. Parkinson Study Group. N Engl J Med 2004; 351: 2498–2508. 10. Olanow et al. Lancet Neurol 2006; 5:1013–1020. *UPDRS Parts II and III Schapira & Obeso. Ann Neurol 2006; 59 (3): 559; Parkinson Study Group. N Engl J Med 1993; 328: 176; Parkinson Study Group. Ann Neurol 1996; 40: 99; Shults et al. Arch Neurol 2002; 59: 1541; Parkinson Study Group. Arch Neurol 2002; 59: 1937; Parkinson Study Group. N Engl J Med 2004; 351: 2498; Olanow et al. Lancet Neurol 2006; 5:1013 23
: 559– Morrish PK, Sawle GV, Brooks DJ. Brain 1996; 119: 585– Fearnley JM, Lees AJ. Brain 1991; 114: 2283– Hilker R, Schweitzer K, Coburger S, et al. Arch Neurol 2005; 62: 378 – Parkinson Study Group. N Engl J Med 1993; 328: 176– Parkinson Study Group. Ann Neurol 1996; 40: 99– Shults et al. Arch Neurol 2002; 59: 1541– Parkinson Study Group. Arch Neurol 2002; 59: 1937– Parkinson Study Group. N Engl J Med 2004; 351: 2498– Olanow et al. Lancet Neurol 2006; 5:1013–1020. *UPDRS Parts II and III. Schapira & Obeso. Ann Neurol 2006; 59 (3): 559; Parkinson Study Group. N Engl J Med 1993; 328: 176; Parkinson Study Group. Ann Neurol 1996; 40: 99; Shults et al. Arch Neurol 2002; 59: 1541; Parkinson Study Group. Arch Neurol 2002; 59: 1937; Parkinson Study Group. N Engl J Med 2004; 351: 2498; Olanow et al. Lancet Neurol 2006; 5:")
24
Follow-up period (months)
PD-LIFE: multizentrische prospektive Studie, on-going, n=198 60 Treatment-naïve patients 50 Monotherapy with any anti-PD drug 40 PDQ-39 single index Deterioration 30 20 The issue of when to start treatment in PD remains controversial.1 Some physicians favour treatment at the time of diagnosis while others opt for a ‘wait and watch’ policy.1 The effect of the latter policy on the health status of people with PD is, to date, unknown.1 The PD-LIFE study was a multicentre, prospective, ‘real-life’ observation audit, conducted to compare self-reported health status (PDQ-39) in patients with PD who were left untreated with those who received treatment.1 A total of 198 untreated patients with PD were enrolled in the study and assessed over a mean period of 18 months.1 Patients were asked to complete a PDQ-39 questionnaire at baseline, 9 months, and 18 months after study entry, to assess the self-reported health status of patients.1 In patients left untreated, the overall PDQ-39 summary index worsened significantly when assessed at 9 and 18 months (p<0.01). However, patients in the comparative group where treatment was initiated at or soon after diagnosis, showed a trend towards improvement in self-reported health status scores after treatment was started.1 Therefore, the PD-LIFE study found that patients with PD, who are treatment-naïve, have significantly worse quality of life than those receiving treatment.1 1. Grosset D, Taurah L, Burn DJ. J Neurol Neurosurg Psychiatry 2007; 78: 465–469. 10 Baseline 9 18 Follow-up period (months) Treatment-naïve PD patients have significantly worse QoL than those receiving treatment Grosset. et al JNNP 2007; 78 (5): 465 24
 in patients with PD who were left untreated with those who received treatment.1. A total of 198 untreated patients with PD were enrolled in the study and assessed over a mean period of 18 months.1. Patients were asked to complete a PDQ-39 questionnaire at baseline, 9 months, and 18 months after study entry, to assess the self-reported health status of patients.1. In patients left untreated, the overall PDQ-39 summary index worsened significantly when assessed at 9 and 18 months (p<0.01). However, patients in the comparative group where treatment was initiated at or soon after diagnosis, showed a trend towards improvement in self-reported health status scores after treatment was started.1. Therefore, the PD-LIFE study found that patients with PD, who are treatment-naïve, have significantly worse quality of life than those receiving treatment Grosset D, Taurah L, Burn DJ. J Neurol Neurosurg Psychiatry 2007; 78: 465– Baseline Follow-up period (months) Treatment-naïve PD patients have significantly worse QoL than those receiving treatment. Grosset. et al. JNNP 2007; 78 (5):")
25
Timing of Treatment Initiation in PD:
A Need for Reappraisal? AHV Schapira, J Obeso 2006 Early correction of the basal ganglia funtional abnormalities caused by dopaminergic cell loss and dopamine deficiency is a means to support the intrinsic physiological compensatory mechanisms DATATOP, Padberg Study, ELLDOPA, TEMPO, ADAGIO Studies

26
Adhärenz Grosset et al. (2009)
")
27
Phasische Stimulation
Levodopa Pre-synaptic membrane Dopamine In the healthy striatum, and to a lesser extent, early on during Parkinson’s disease, dopamine can be stored in the striatal neurones and released when needed.1,2 In the early stages of Parkinson’s disease there are still adequate numbers of residual dopamine terminals in the brain to store the dopamine converted from levodopa and buffer fluctuations in plasma levodopa concentration. This permits relatively continuous and physiological release of dopamine and stimulation of dopamine receptors.1 However, as the disease progresses, when short-acting medications, such as levodopa, are administered intermittently, intrasynaptic dopamine levels begin to vary. The large shifts in levodopa concentrations lead to large variation in dopamine levels and dopaminergic activity at the synapse.1,3 When levodopa is taken up into degenerating neurones, the release of dopamine is accelerated.4 As the number of surviving dopaminergic terminals degenerates, regulatory mechanisms are lost and dopamine levels fluctuate widely.4,5 Increasing the frequency of dosing in order to minimise the wearing off that develops between doses is successful to some extent at first, but does not address the pulsatile delivery of levodopa, and large fluctuations in plasma drug levels remain.6 References 1. Olanow CW et al. Neurology 2001;56 (Suppl 5):S1–S88. 2. Thanvi BR et al. Postgrad Med J 2004;80:452–8. 3. Stocchi F et al. Neurology 2004;62 (Suppl 1):S56–S63. 4. Chase TN et al. Adv Neurol 1996;69:497–501. 5. Abercrombie ED et al. Brain Res 1990; 525:36–44. 6. Olanow CW et al. Nat Clin Pract Neurol 2006;2:382–92. Post-synaptic membrane Adapted from Thanvi BR et al. Postgrad Med J 2004;80:452–8.
:S1–S Thanvi BR et al. Postgrad Med J 2004;80:452–8. 3. Stocchi F et al. Neurology 2004;62 (Suppl 1):S56–S Chase TN et al. Adv Neurol 1996;69:497– Abercrombie ED et al. Brain Res 1990; 525:36– Olanow CW et al. Nat Clin Pract Neurol 2006;2:382–92. Post-synaptic membrane. Adapted from Thanvi BR et al. Postgrad Med J 2004;80:452–8.")
28
Beginn mit einem MAO-B-Hemmer
Jenner P & Langston JW (2011) Movement Disord 26: RASAGILIN
 Movement Disord 26: RASAGILIN.")
29
Delayed-start Design Symptomatischer vs. Krankheitsmod. Effekt
Plazebo Delayed-start Symptomatisch Early-start Spättherapierte holen auf Symptom Verbesserung Zeit Zeit Spättherapierte holen nicht auf Symptom Verbesserung Symptomatisch & Krankheitsmod. Plazebo Delayed-start Early-start In the delayed-start design, some patients are randomised to receive the investigational agent immediately (early-start), while others are randomised to begin after a delay (delayed-start).1,2 The trial design presumes that the symptomatic benefit of the medication is similar in both groups at the end of treatment.2,3 Any difference between early- and delayed-start, over and above a symptomatic benefit, must be due to a disease-modifying effect.1,2 A drug which has symptomatic effects only will show the same maximal benefit no matter when, in the course of the disease, the drug is started – early or delayed. However, if a drug possesses both symptomatic and disease-modifying effects, introducing the active treatment to the delayed-start patients produces an initial symptomatic benefit, after which point a deterioration is observed. A the end of the trial, a greater benefit is achieved when the drug is started earlier (early-start), resulting in less functional impairment over time. Since the design of the delayed-start trial presumes that any symptomatic effect should be equal in the two groups at the end of the trial, the difference must be due to earlier treatment having a disease-modifying effect.2 1. Leber P. Alzheimer Dis Assoc Disord 1997; 11 (5): S10–S21. 2. Clarke CE. Mov Disord 2008; 23 (6): 784–789. 3. Hung AY, Schwarzschild MA. Curr Opin Neurol 2007; 20: 477–483. 29
, while others are randomised to begin after a delay (delayed-start).1,2. The trial design presumes that the symptomatic benefit of the medication is similar in both groups at the end of treatment.2,3 Any difference between early- and delayed-start, over and above a symptomatic benefit, must be due to a disease-modifying effect.1,2. A drug which has symptomatic effects only will show the same maximal benefit no matter when, in the course of the disease, the drug is started – early or delayed. However, if a drug possesses both symptomatic and disease-modifying effects, introducing the active treatment to the delayed-start patients produces an initial symptomatic benefit, after which point a deterioration is observed. A the end of the trial, a greater benefit is achieved when the drug is started earlier (early-start), resulting in less functional impairment over time. Since the design of the delayed-start trial presumes that any symptomatic effect should be equal in the two groups at the end of the trial, the difference must be due to earlier treatment having a disease-modifying effect Leber P. Alzheimer Dis Assoc Disord 1997; 11 (5): S10–S Clarke CE. Mov Disord 2008; 23 (6): 784– Hung AY, Schwarzschild MA. Curr Opin Neurol 2007; 20: 477–")
30
TEMPO 12-Monatsergebnisse: Mittlere Änderung im UPDRS-Total
Primäre Analyse: 371 Personen -2 -1 1 2 3 4 UPDRS-Total Veränderung Rasagilin 1 mg/Tag Rasagilin 2 mg/Tag Verspätet 2 mg/Tag Rasagilin Woche * Delayed start ** 14 20 52 8 42 26 32 *p=0.05 **p=0.01 Efficacy analysis of the TEMPO 12-month study period was performed on 371 subjects who had at least one UPDRS measurement in the active treatment phase before receiving additional anti-PD therapy. At the end of the 52-week study, 259 subjects were still receiving rasagiline monotherapy. The treatment effect with regard to the primary outcome measure, UPDRS-Total score, observed in the first 6 months persisted at 12 months, such that patients treated with rasagiline for all 12 months showed less decline in UPDRS scores than patients whose active treatment was delayed for 6 months.1,2 1. Parkinson Study Group. Arch Neurol 2002; 59: 1937–1943. 2. Parkinson Study Group. Arch Neurol 2004; 61: 561–566. Parkinson Study Group. Arch Neurol 2002; 59: 1937; Parkinson Study Group. Arch Neurol 2004; 61: 561 30

31
TEMPO Langzeit-Beobachtungsstudie
* *** *p<0.05 ***p<0.001 Overall difference between early and delayed start groups (repeated measures analysis) is 16% (p=0.006) (n=404) (n=324) (n=272) (n=237) (n=206) (n=164) Years Improvement 2.5 unit difference Placebo vs rasagiline phase For the entire treatment period of the TEMPO study (up to 6.5 years), the mean difference in the change from baseline in UPDRS-Total between the early and delayed rasagiline treatment groups after 6.5 years was 2.5 ± 1.1 (p=0.0206).1 This corresponds to a mean difference in percent change from baseline between groups of 16 ± 5.7% (p=0.006).1 When data were compared for each half-year interval, significantly less symptom progression and functional decline were seen in the early treatment group at the 0.5, 1.5, 2.0, 3.0, 4.5, 5.0, and 5.5 year time points (p<0.05).1 (On the graph, the 6 year and 6.5 year data points have been combined.) 1. Hauser RA, Lew MF, Hurtig HI, et al, and the TEMPO Extension Study Group. Poster presented at 16th ICPD. Berlin, Germany. June 2005. Data points for 6 and 6.5 years are combined Hauser et al. Poster at ICPD, 2005 Hauser et al. Park and Rel Dis 2005, 11 (Suppl. 2), 129 31
 is 16% (p=0.006) (n=404) (n=324) (n=272) (n=237) (n=206) (n=164) Years. Improvement. 2.5 unit difference. Placebo vs rasagiline phase. For the entire treatment period of the TEMPO study (up to 6.5 years), the mean difference in the change from baseline in UPDRS-Total between the early and delayed rasagiline treatment groups after 6.5 years was 2.5 ± 1.1 (p=0.0206).1 This corresponds to a mean difference in percent change from baseline between groups of 16 ± 5.7% (p=0.006).1. When data were compared for each half-year interval, significantly less symptom progression and functional decline were seen in the early treatment group at the 0.5, 1.5, 2.0, 3.0, 4.5, 5.0, and 5.5 year time points (p<0.05).1. (On the graph, the 6 year and 6.5 year data points have been combined.) 1. Hauser RA, Lew MF, Hurtig HI, et al, and the TEMPO Extension Study Group. Poster presented at 16th ICPD. Berlin, Germany. June Data points for 6 and 6.5 years are combined. Hauser et al. Poster at ICPD, Hauser et al. Park and Rel Dis 2005, 11 (Suppl. 2),")
32
361(13): , September 24, 2009
: , September 24, 2009")
33
ADAGIO: Studien Design
1 mg/Tag Frühe, unvorbehandelte Parkinson-Patienten (N=1,176) 1 mg/Tag Plazebo 2 mg/Tag 2 mg/Tag Randomisation 1:1:1:1 Woche -4 -4 4 12 24 36 42 48 54 60 66 72 36wöchige doppelblinde Verum-Behandlungsphase 36wöchige doppelblinde Plazebo-kontrollosierte Phase
 1 mg/Tag. Plazebo. 2 mg/Tag. 2 mg/Tag. Randomisation 1:1:1:1. Woche wöchige doppelblinde Verum-Behandlungsphase. 36wöchige doppelblinde. Plazebo-kontrollosierte Phase.")
34
Figure 3. Changes in Scores on the Unified Parkinson’s Disease Rating Scale (UPDRS) in the Four Study Groups. The mean (±SE) change from baseline in the UPDRS score in the efficacy cohort for the second and third primary end points for patients receiving rasagiline at a dose of 1 mg per day (Panel A) and those receiving 2 mg per day (Panel B) are shown. The dashed lines indicate placebo, and the solid lines indicate rasagiline.
 change from baseline in the UPDRS score in the efficacy cohort for the second and third primary end points for patients receiving rasagiline at a dose of 1 mg per day (Panel A) and those receiving 2 mg per day (Panel B) are shown. The dashed lines indicate placebo, and the solid lines indicate rasagiline.")
35
‘Floor’-Effekt in der UPDRS-Skala
Niedrigere UPDRS vor Behandlungsbeginn KM Wirkung ist nicht nachweisbar Zeit Nur symptomatisch (Potenzielle Verbesserung von X UPDRS-Einheiten) Nur symptomatisch + KM von X+Y UPDRS-Einheiten) Floor-Effekt X Y Arzneimittel Nur symptomatisch (Potenzielle Verbesserung von X UPDRS-Einheiten) Symptomatisch + KM von X+Y UPDRS-Einheiten) Zeit Höhere UPDRS vor Behandlungsbeginn KM Wirkung ist nachweisbar X Y Arzneimittel Verschlechterung Verbesserung Das Schaubild auf der linken Seite verdeutlicht, warum bei Patienten mit milderer Erkrankung und geringeren UPDRS-Scores vor Behandlungsbeginn möglicherweise keine zusätzlichen krankheitsmodifizierenden Wirkungen (Y) nachgewiesen werden können. Wenn die symptomatischen Wirkungen (X) des Arzneimittels bereits die maximal wahrnehmbaren Nutzen in den UPDRS-Scores erzielen (niedrigst-möglicher Score), so könnte dieser ‘Floor’-Effekt erklären, warum die gesamte Kohorte (mittlerer UPDRS-Gesamtscore vor Behandlungsbeginn von 20) keine signifikante krankheitsmodifizierende Wirkung mit Rasagilin 2 mg/Tag gezeigt hat. Das Schaubild auf der rechten Seite verdeutlicht, wie es höhere UPDRS-Scores vor Behandlungsbeginn ermöglichen, die gleichen symptomatischen und krankheitsmodifizierenden Wirkungen eines Arzneimittels wie im rechten Schaubild leichter zu erkennen. Der ‘Floor’-Effekt kann erklären, warum im oberen Quartil von Rasagilin 2 mg/Tag – der Subgruppe von Patienten mit UPDRS-Gesamtscores von ≥25,5 vor Behandlungsbeginn – ein signifikanter Nutzen beobachtet wurde (also nachweisbar war). KM=krankheitsmodifizierend
 Nur symptomatisch + KM. von X+Y UPDRS-Einheiten) Floor-Effekt. X. Y. Arzneimittel. Nur symptomatisch. (Potenzielle Verbesserung. von X UPDRS-Einheiten) Symptomatisch + KM. von X+Y UPDRS-Einheiten) Zeit. Höhere UPDRS vor Behandlungsbeginn KM Wirkung ist nachweisbar. X. Y. Arzneimittel. Verschlechterung. Verbesserung. Das Schaubild auf der linken Seite verdeutlicht, warum bei Patienten mit milderer Erkrankung und geringeren UPDRS-Scores vor Behandlungsbeginn möglicherweise keine zusätzlichen krankheitsmodifizierenden Wirkungen (Y) nachgewiesen werden können. Wenn die symptomatischen Wirkungen (X) des Arzneimittels bereits die maximal wahrnehmbaren Nutzen in den UPDRS-Scores erzielen (niedrigst-möglicher Score), so könnte dieser ‘Floor’-Effekt erklären, warum die gesamte Kohorte (mittlerer UPDRS-Gesamtscore vor Behandlungsbeginn von 20) keine signifikante krankheitsmodifizierende Wirkung mit Rasagilin 2 mg/Tag gezeigt hat. Das Schaubild auf der rechten Seite verdeutlicht, wie es höhere UPDRS-Scores vor Behandlungsbeginn ermöglichen, die gleichen symptomatischen und krankheitsmodifizierenden Wirkungen eines Arzneimittels wie im rechten Schaubild leichter zu erkennen. Der ‘Floor’-Effekt kann erklären, warum im oberen Quartil von Rasagilin 2 mg/Tag – der Subgruppe von Patienten mit UPDRS-Gesamtscores von ≥25,5 vor Behandlungsbeginn – ein signifikanter Nutzen beobachtet wurde (also nachweisbar war). KM=krankheitsmodifizierend.")
36
PROUD: Pramipexole Early-Start Study
UPDRS SPECT QoL-CGI UPDRS CGI UPDRS CGI UPDRS QoL-CGI UPDRS SPECT QoL-CGI PPX 1.5 mg PPX 1.5 mg Early untreated PD Placebo 3m 6m 9m 15m Titration 6wk Titration 6wk

37
Protektiver Effekt von Cabergolin auf die Morphologie
control 80 nM rotenone 48 h 1.: 0.5 nM cab. 24 h 2.: 80 nM rot. 48 h Protektiver Effekt von Cabergolin auf die Morphologie Gille et al. 2006

38
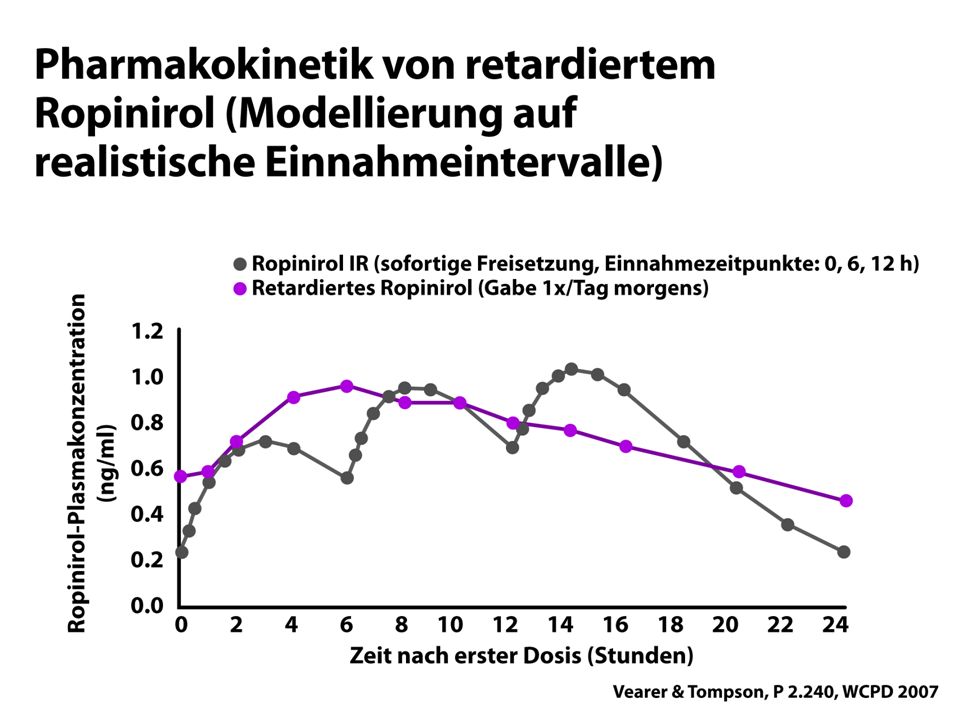
Ropinirol PR

40
Monotherapie-Studie ReQuip ReQuip-MODUTAB Abnahme UPDRS Motor Score
n.s.

41
Aufbau der Hydrogel Matrix

42
Signifikante Reduktion des UPDRS II + III nach 18 Wochen
Pramipexol Retard - Parkinson im Frühstadium Verlauf UPDRS II + III – Score über 18 Wochen Gezeigt wird die Entwicklung des UPDRS-II+III-Scores (Unified Parkinson´s Disease Rate Scale – UPDRS) über den 18-wöchigen Studienverlauf. Es konnte ein statistisch signifikanter Rückgang dieser Scores für Pramipexol Retard und Pramipexol im Vergleich zu Placebo beobachtet werden. Interessant ist, dass der UPDRS-II+III-Score für Pramipexol Retard und Pramipexol zu jedem Zeitpunkt nahezu identisch war. Literatur 1. Hauser R, Salin L, Koester J. Double-blind evaluation of pramipexole extended-release (ER) in early Parkinson's disease. 61st Ann Mtg of the American Academy of Neurology (AAN), Seattle, 25 Apr - 2 May 2009 (Oral Presentation) 2009; .. Signifikante Reduktion des UPDRS II + III nach 18 Wochen Hauser R, et al, American Academy of Neurology 61st Annual Meeting, Seattle, Washington, USA, April 25-May 2, 2009, Platform S 42
 über den 18-wöchigen Studienverlauf. Es konnte ein statistisch signifikanter Rückgang dieser Scores für Pramipexol Retard und Pramipexol im Vergleich zu Placebo beobachtet werden. Interessant ist, dass der UPDRS-II+III-Score für Pramipexol Retard und Pramipexol zu jedem Zeitpunkt nahezu identisch war. Literatur. 1. Hauser R, Salin L, Koester J. Double-blind evaluation of pramipexole extended-release (ER) in early Parkinson s disease. 61st Ann Mtg of the American Academy of Neurology (AAN), Seattle, 25 Apr - 2 May 2009 (Oral Presentation) 2009; .. Signifikante Reduktion des UPDRS II + III nach 18 Wochen. Hauser R, et al, American Academy of Neurology 61st Annual Meeting, Seattle, Washington, USA, April 25-May 2, 2009, Platform S")
43
RECOVER-Studie -1 -2 -3 -4 -5 -6 -7 -8 p<0,0001
Morgendliche Bewegungsstörungen (UPDRS III) und Schlafqualität (PDSS-2) UPDRS III PDSS-2 1,9 -1 (± 8,2) -2 3,9 -3 (± 7,3) Rotigotin (n=191) Plazebo (n=96) Änderungen im UPDRS III bzw. PDSS-2 (FAS/LOCF) Verbesserung -4 5,9 -5 7,0 (± 7,6) -6 (± 7,6) -7 -3,55 (LS-Mean) p=0,0002 - p<0,0001 -8 16 Trenkwalder C. et al. ^Movement Disorders 2011
 und Schlafqualität (PDSS-2) UPDRS III. PDSS-2. 1, (± 8,2) -2. 3, (± 7,3) Rotigotin (n=191) Plazebo (n=96) Änderungen im UPDRS III. bzw. PDSS-2 (FAS/LOCF) Verbesserung , ,0. (± 7,6) -6. (± 7,6) ,55 (LS-Mean) p=0, p<0, Trenkwalder C. et al. ^Movement Disorders")
44
Paus et al. MovDisord 2003;18:659-667
Schlafattacken bei Einnahme von DA Paus S et al. (2007) Movement Disord 18: Paus et al. MovDisord 2003;18:
 Movement Disord 18: Paus et al. MovDisord 2003;18:")
45
Patientin mit Kaufsucht
Es begann bei mir mit tausenden kreativen Ideen, ich renovierte, trotz der massiven körperlichen Einschränkungen Stück für Stück unser Haus von oben bis unten. Nach jeder Aktion war ich fix und fertig. Alles wurde umdekoriert und, was das Schlimmste war, ich kaufte und kaufte. Sicher, alles schöne Dinge, aber eigentlich zum größten Teil überhaupt nicht notwendig. Ich konnte durch kein Geschäft gehen, ohne irgendetwas zu sehen, was ich meiner Meinung unbedingt benötigte. Nachts schlief ich immer weniger, manche Nächte blieb ich ganz wach, es stellte sich kein Schlafbedürfnis ein. Setzte man mich aber z. B. (als Beifahrer) ins Auto, schlief ich sofort ein. Überall hatte ich, wie mein Mann es nannte, meine Baustellen aufgebaut und sprang zwischen ihnen hin und her. Ich litt unter Appetitlosigkeit, aß eigentlich nur noch aus Vernunft und nahm natürlich dadurch auch einiges an Gewicht ab. Meinen Gemütszustand möchte ich fast als manisch beschreiben. Mein Mann redete immer wieder mit mir, aber ich nahm nicht wirklich etwas an. Natürlich war mir klar, dass diese Einkauferei nicht ok ist, aber ich fand für mich immer eine Entschuldigung.
 ins Auto, schlief ich sofort ein. Überall hatte ich, wie mein Mann es nannte, meine Baustellen aufgebaut und sprang zwischen ihnen hin und her. Ich litt unter Appetitlosigkeit, aß eigentlich nur noch aus Vernunft und nahm natürlich dadurch auch einiges an Gewicht ab. Meinen Gemütszustand möchte ich fast als manisch beschreiben. Mein Mann redete immer wieder mit mir, aber ich nahm nicht wirklich etwas an. Natürlich war mir klar, dass diese Einkauferei nicht ok ist, aber ich fand für mich immer eine Entschuldigung.")
46
Abhängigkeiten: Beispiele
- Pathologische Spielsucht - Hypersexualität - Hobbyismus - Punding - Suchthaftes Einkaufen - Medikamentensucht - Rücksichtloses Autofahren - Fressen u.a.

47
Impulskontrollstörungen beim IPS Prävalenz
Weintraub et al. 2006 ICS-Prävalenz 6,6 % Voon et al. 2006 ICS-Prävalenz total 6,1% DA-agonist 13,7% Normalkollektiv 0,4-1%

48
Punding Characterisierung:
Repetitive, nutzlose Bewegungen wie Sammeln, Arrangieren, oder Auseinanderbauen von Gegenständen oder dauerndes an sich Herumzupfen. (Bisher kannte man dieses Phänomen nur von Patienten, die einen Kokain oder Amphethamin-Missbrauch betrieben (Dopamin Überdosis)
")
49
Behandlung in der Frühphase
Diagnosis Decision to refer to neurologist Decision to treat Yes Evaluate patient characteristics and degree of disability Mild motor disability and no cognitive impairment Moderate/Severe motor disability and no cognitive impairment F Moderate/severe disability and age 70–75+ years or with significant co-morbidity including cognitive impairment Reference Schapira AH. Treatment options in the modern management of Parkinson disease. Arch Neurol 2007;64(8): Begin dopamine agonist or MAO-B inhibitor Begin dopamine agonist Begin levodopa* Schapira AH. Arch Neurol 2007;64(8):
: Begin dopamine agonist or MAO-B inhibitor. Begin dopamine agonist. Begin levodopa* Schapira AH. Arch Neurol 2007;64(8):")
50
Danke für die Aufmerksamkeit, besuchen Sie uns in Dresden!

Ähnliche Präsentationen














 Schwester denn zum Geburtstag?>")
 András Bárdossy IWS Universität Stuttgart.>")



