Präsentation herunterladen
Die Präsentation wird geladen. Bitte warten

1
A probabilistic model for the evolution of RNA structure – Holmes
RNA secondary structure prediction with simple pseudoknots – Deogun, Donis, Komina, Ma

2
Gliederung Einführung Das TKF91 Modell Der TKF91 Structure Tree
Vorstellung der durchgeführten Tests Interpretation der Ergebnisse

3
Ergebnis des Humangenomprojekts:
„Gut DREI Prozent brauchbares Material und jede Menge Schrott.“ Vergleich Genomgröße mit #Genen RNA kann mehr als genetische Infomration von A nach B zu tragen. Genetische Regulation, katalytische Funktion, Regulation zellulärer Prozesse

4
snRNA Splicing von pre-mRNA guideRNA RNA-Editing in Mitchondrien
Ribonukleasen Regulation der Biosynthese von tRNA tRNA Proteinbiosynthese rRNA Proteinbiosynthese Telomerase RNA DNA Synthese an chromsomalen Enden snoRNA Methylierung von rRNA Non-coding RNA kann nicht durch ORF Vorhersage gefunden werden

5
Identifikation funktioneller Signale in einer Gensequenz. Idee:
Ziel: Identifikation funktioneller Signale in einer Gensequenz. Idee: Funktionelle Signale sind evolutionär konserviert. Vorgehensweise: Fitten der Daten an probabilistische Modelle, die den evolutionären Prozess darstellen.

6
Es existieren verschiedene Arten von konservierten Elementen x, y, z…
Für jedes Szenario kann man ein probabilistisches Modell Mx, My, Mz erstellen. Die Likelihood der beobachteten Daten unter jedem dieser Modelle werden verglichen. Modell mit der besten Anpassung zeigt den Typ des funktionellen Elements.

7
Es existieren zwei Vorgehensweisen zur Verwendung von evolutionärer
Distanz: Trainingsalignments werden eingeteilt nach ihrer prozentualen Sequenz- identität. Alignments, die gleich eingeteilt wurden, repräsentieren dann Sequenzen mit äquivalenten Distanzen. (siehe BLOSUM) Evolutionäre Distanz wird als Zeitmessung betrachet. Man legt einen stochastischen Prozess zugrunde, mit konstanten Mutationsparametern. (siehe PAM) 2. Ansatz im Kontext der der likelihood-basierenden Methoden der Phylogenie besser, da kompatibler
 Evolutionäre Distanz wird als Zeitmessung betrachet. Man legt einen. stochastischen Prozess zugrunde, mit konstanten Mutationsparametern. (siehe PAM) 2. Ansatz im Kontext der der likelihood-basierenden Methoden der Phylogenie besser, da kompatibler")
8
Bisherige Ansätze zur Identifikation von funktioneller non-coding RNA
betrachten Sekundärstruktur nicht. Aber: Funktion und Struktur sind eng miteinander verknüpft In der Biologie ist Funktion immer bedingt durch Struktur Daher: Neues Modell betrachtet evolutionäre Entwicklung von Sekundärstruktur

9
Gliederung Einführung Das TKF91 Modell Der TKF91 Structure Tree
Vorstellung der durchgeführten Tests Interpretation der Ergebnisse

10
Einfluss von 2 Arten von Mutations-Ereignissen:
TKF91-Modell beschreibt die Evolution einer einzelnen Sequenz unter dem Einfluss von 2 Arten von Mutations-Ereignissen: 1. Punkt-Substitutionen 2. InDel-Ereignisse Die Raten der Mutations-Ereignisse sind unabhängig von benachbarten Ereignissen.

11
Das Modell ist zeit-reversibel
Es kann oBdA davon ausgegangen werden, dass eine der beiden Sequenzen die Ursequenz der anderen ist.

12
Punkt - Substitutionen
Positionen evolvieren unabhängig voneinander Zugrunde liegendes Substitionsmodell fij(t) = WS für Übergang von Base i nach j zur Zeit t s = Rate der Basensubstitution
 = WS für Übergang von Base i nach j zur Zeit t. s = Rate der Basensubstitution")
13
Insertionen - Deletionen
A G C U U A C C G A N+1 Positionen, an denen eingefügt werden kann - mit Rate N Positionen, an denen gelöscht werden kann - mit Rate < vorrausgesetzt kein Ungleichgewicht immortal link mortal links Jede Position = Markov Kette mit 4 Zuständen, auf der diese herumspringt. Gedächtnislosigkeit => neues Nukleotid nur vom letzten abhängig # überlebender Nachkommen ist geometrisch verteilt (wenn Urposition überlebt)
")
14
Folgende Wahrscheinlichkeiten ergeben sich aus Raten n und n:
n = Wahrscheinlichkeit einer Nicht-Deletion n = Wahrscheinlichkeit einer Insertion n = Wahrscheinlichkeit einer Insertion nach einer Deletion n = Wahrscheinlichkeit die Sequenz fortzuführen Außerdem ist Mn(i,j) die Substitutionswahrscheinlichkeit von Base i durch j
 die Substitutionswahrscheinlichkeit von Base i durch j")
15
Die Sequenzlänge im Gleichgewicht ist geometrisch verteilt,
mit Parameter. Sequenzlänge im Gleichgewicht ist geometrisch verteilt mit Parameter Kappa0

17
Gliederung Einführung Das TKF91 Modell Der TKF91 Structure Tree
Vorstellung der durchgeführten Tests Interpretation der Ergebnisse

18
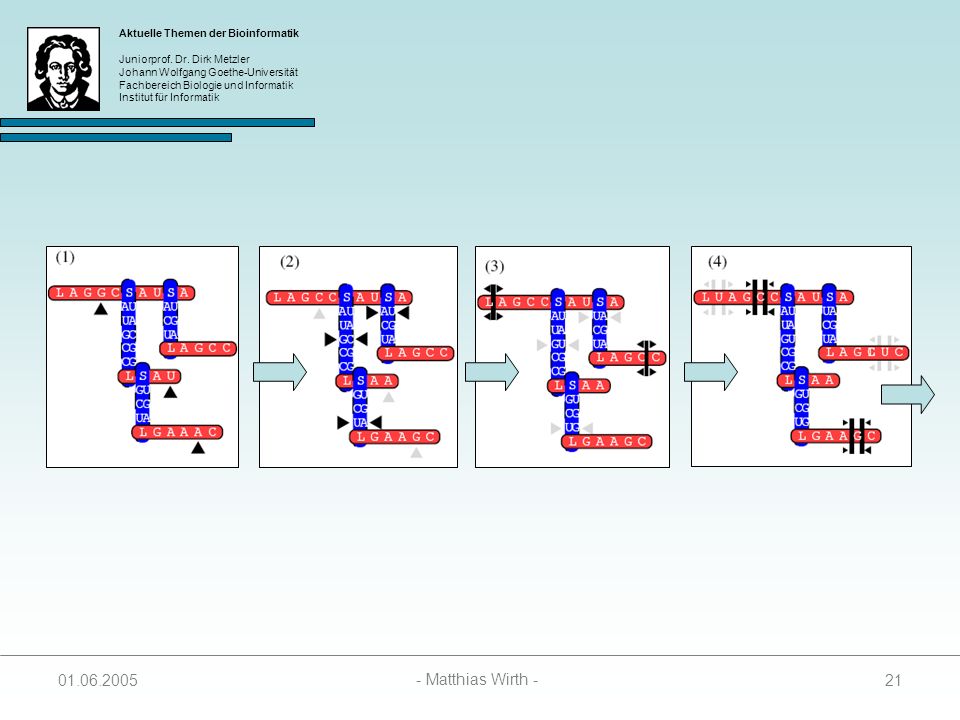
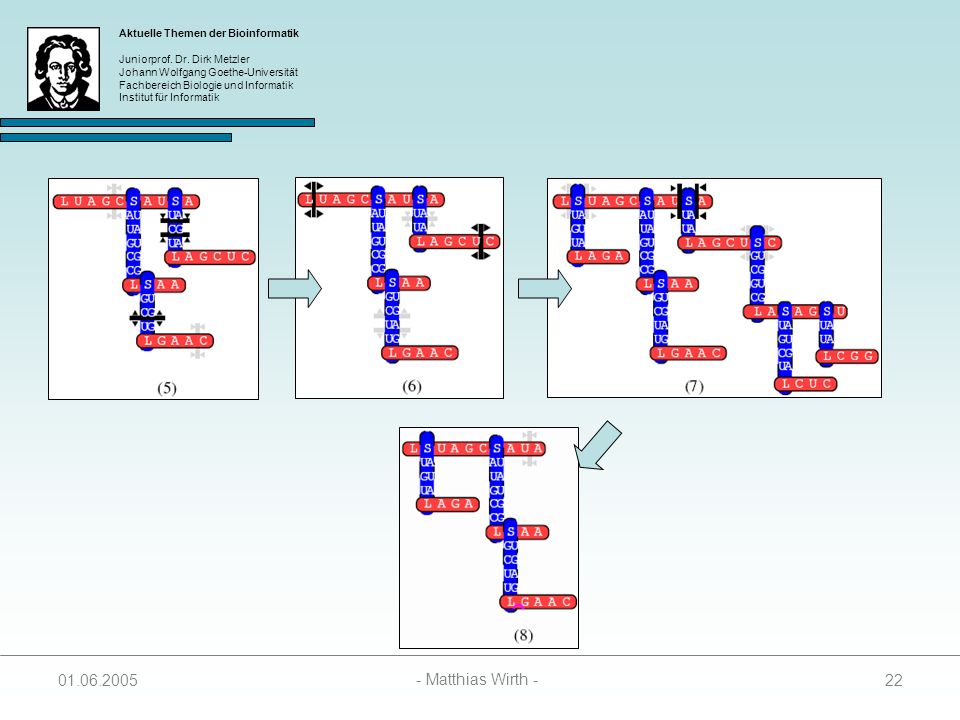
Gewurzelter Baum, indem jeder Knoten einen Grad 3 besitzt.
Beschreibt die Sekundärstruktur einer RNA-Sequenz 4 Arten von Knoten: 1. singlet: 2. paired: 3. loop: 4. stem: Struktur wird vom Auftreten von Loop- und Stem-Knoten bestimmt unabhängig evolvierende Nukleotide kovariante Basenpaare Anfang einer Loopsequenz Anfang einer Stemsequenz

19
Knotenbeschriftungen:
= { L, S } = { A, C, G, U } ² = { AA, AC, AG, AU, CA, CC, CG, CU, GA, GC, GG, GU, UA, UG, UC, UU }

20
R1(X, S) = R1(S,X) = 0 für alle X element Omega

24
Implementierung der Grammatik-Parser setzt eine Umgestaltung der
Grammatik voraus. Problemstellen: Null-Zykel - können durch Loop-/Stemlängen = 0 entstehen Silent Bulges - S S Loop Bifurcation - L LL

25
Null-Zykel (4, 7, 11) Loop Bifurcation (24, 27) Silent Bulge
(32, 29, 30)
")
27
Komplexität der Algorithmen:
single sequence SCFG: Zeit: (L³) Platz: (L²) pairwise SCFG: Zeit: (L³M³) Platz: (L²M²) => Finden des wahrscheinlichsten Parse-Baums mit Hilfe des CYK-Algorithmus
 Platz: (L²) pairwise SCFG: Zeit: (L³M³) Platz: (L²M²) => Finden des wahrscheinlichsten Parse-Baums mit Hilfe des CYK-Algorithmus")
28
Gliederung Einführung Das TKF91 Modell Der TKF91 Structure Tree
Vorstellung der durchgeführten Tests Interpretation der Ergebnisse

29
Implementierung eines Alignment-Tool auf Basis der SCFG‘s
Basierend auf dynamischen Programmieren mit beschleunigenden Heuristiken Als Test der Leistungsfähigkeit des Modells werden Paare von RNA-Sequenzen miteinander aligniert und deren Struktur vorhergesagt. 4 verschiedene Familien mit variierender Homologie im Bereich der Sekundärstruktur wurden ausgewählt.

30
summiert über alle Alignments mit der anderen Sequenz
Alignment mit dem TKF91-Structure Tree Strukturvorhersage für einzelne Sequenz Strukturvorhersage summiert über alle Alignments mit der anderen Sequenz Alignment mit dem TKF91-Modell

31
Identische Sekundärstruktur Primärsequenz weicht voneinander ab
Purine Riboswitch Identische Sekundärstruktur Primärsequenz weicht voneinander ab

32
Regulatorische Elemente die spezifisch Adenin oder Guanin binden.
Sind Bestandteil der post-translationalen Regulation von Purin-Transport und –biosynthese.

33
Nano translational control element
Deletion des äußeren Stems

34
Regulatorische Sequenz im 3‘ UTR der nano-Gene von Drosophila.
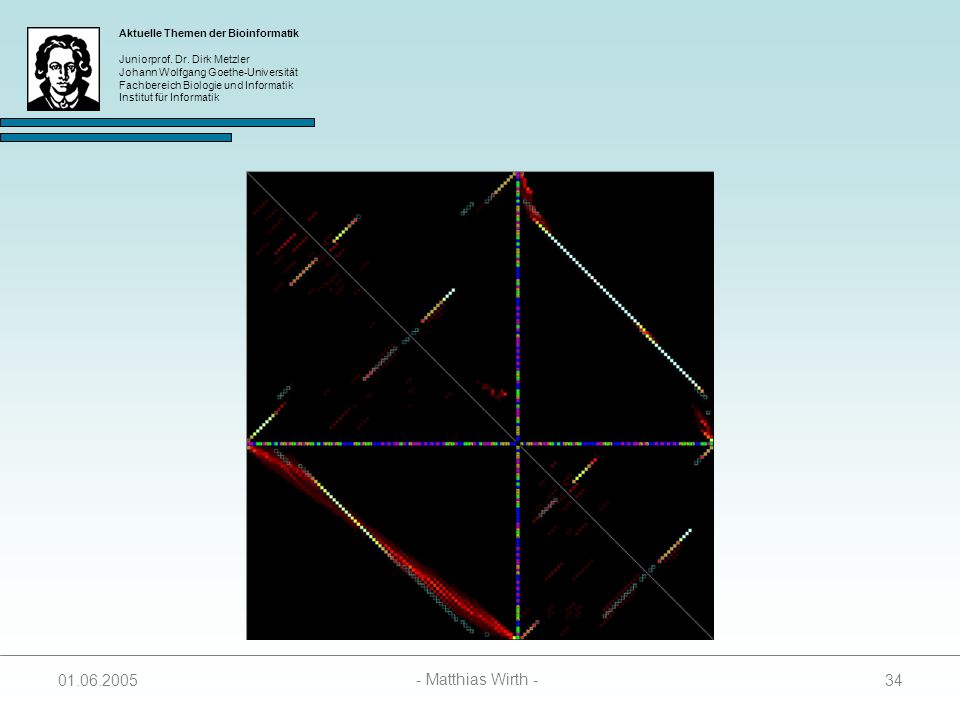
35
sehr ähnliche Primärsequenz
U2 splicing factors Deletion von Stem 4, 5 und 6 sehr ähnliche Primärsequenz

36
Bindet am Schneidepunkt zwischen Exon und Intron und markiert Schnittstelle in pre-mRNA‘s
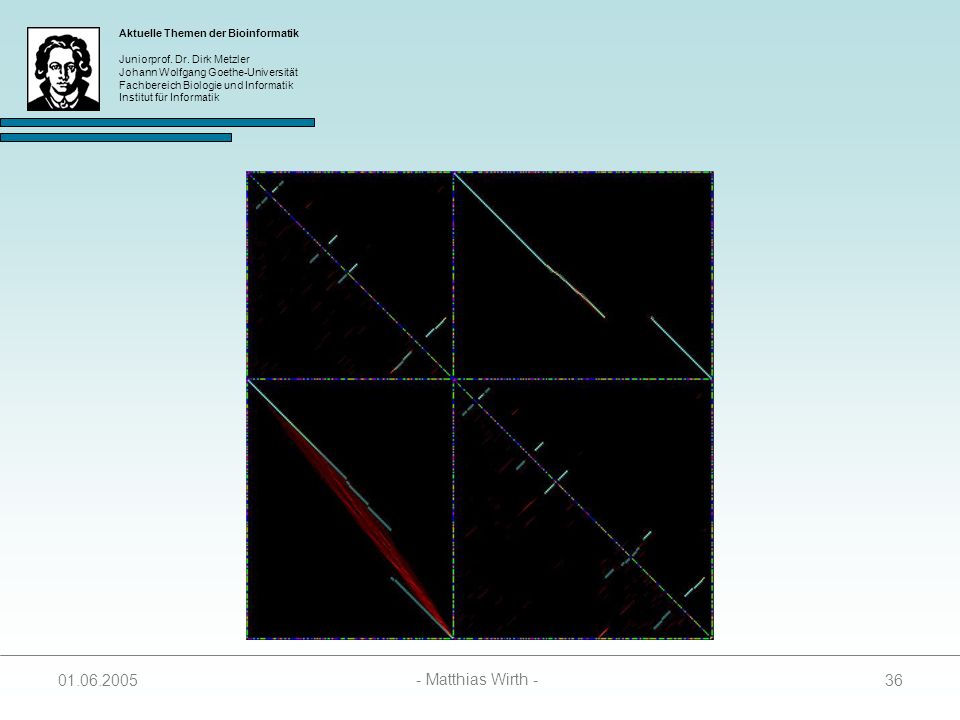
37
in der Sekundärstruktur variabelste Familie in RFAM
RNase P Genes Starker Unterschied in der Sekundärstruktur variabelste Familie in RFAM

38
Endoribonuklease Ribozym (katalytische Funktion)
Spielt Rolle in der Biosynthes von tRNA‘s.

39
StructureTree singlet
RNA Sequenzen Strukturvorhersage StructureTree singlet StructureTree paired pairHMM Alignment pairSCFG Alignment Purine Riboswitches schlecht korrekt Nano translational control element gut Probleme in den Rand-bereichen wesentlich besser U2 splicing factors RNase P

40
Gliederung Einführung Das TKF91 Modell Der TKF91 Structure Tree
Vorstellung der durchgeführten Tests Interpretation der Ergebnisse

41
streng konservierte Struktur und wenige InDels führen zu guter
Stärken des Modells: streng konservierte Struktur und wenige InDels führen zu guter Struktur-Vorhersage und Alignment Bei vielen InDels in Loops und Stems oder bei geringfügigen Änderungen der Sekundärstruktur arbeitet der StructureTree auch gut Schwächen des Modells: Ab einem bestimmten Grad der strukturellen Unterschiede zwischen Sequenzen versagt das Modell (RNase P)
")
42
Mögliche Verbesserungen:
Hinzunahme von „long indels“ und affiner Gap-Penalty zusätzliche Modellierung von Thermodynamik-Effekten (Basepair Stacking, Nearst Neighbour Interaktion) Verbessertes Einfügen von Bulges (Zulassen von L-Knoten in Stems) Annahme das Stems und Loops alle mit der gleichen Rate evolvieren ist empirisch nicht belegt Triloops, Tetraloops, U-Turns u.ä. werden nicht speziell behandelt, obwohl oft evolutionär konserviert Einführung spezieller InDel-Raten für Stems/MultiStems (bislang gleiche Raten) Verbesserung der Stem-Deletion, äußer Stems sollten nicht zwangsläufig zu Löschung von inneren führen. Belegt durch empirische Studien in RFAM.
 Verbessertes Einfügen von Bulges (Zulassen von L-Knoten in Stems) Annahme das Stems und Loops alle mit der gleichen Rate evolvieren ist empirisch nicht belegt. Triloops, Tetraloops, U-Turns u.ä. werden nicht speziell behandelt, obwohl oft evolutionär. konserviert. Einführung spezieller InDel-Raten für Stems/MultiStems (bislang gleiche Raten) Verbesserung der Stem-Deletion, äußer Stems sollten nicht zwangsläufig zu Löschung von. inneren führen. Belegt durch empirische Studien in RFAM")
43
A probabilistic model for the evolution of RNA structure – Holmes
RNA secondary structure prediction with simple pseudoknots – Deogun, Donis, Komina, Ma

44
Gliederung Einleitung Algorithmus von Akutsu
Nearest Neighbour Thermodynamik Regeln Berechnung minimaler Energien von RNA-Substrukturen Optimale Energie eines Pseudoknots Analyse des Algorithmus

45
In diesem Algorithmus werden nur einfache Pseudoknoten betrachtet.
Wie bereits gesehen, ist die Pseudoknoten-Vorhersage kein triviales Problem. Die Möglichkeit zur Vorhersage ist aber wichtig, da Pseudoknoten verbreitete Strukturen sind, die eine wichtige Rolle in funktionell wichtiger RNA spielen. In diesem Algorithmus werden nur einfache Pseudoknoten betrachtet. Algorithmus wurde entwickelt, um Sequenzen mit Länge >100 betrachten zu können. Laufzeitverbesserung im Gegensatz zu Eddy/Rivas: ER = (n6) Zeit, (n4) Platz DK = (n4) Zeit, (n3) Platz
 Zeit, (n4) Platz DK = (n4) Zeit, (n3) Platz")
46
zur Vorhersage von Pseudoknots
Nearest Neighbour Thermodynamik Regeln mfold-Algorithmus Neuer Algorithmus zur Vorhersage von Pseudoknots Akutsu-Algorithmus zur Vorhersage von Pseudoknots unter Maximierung von Basenpaaren

47
Eine Sekundärstruktur S einer RNA-Sequenz A = a1a2…an ist eine
Definition: Eine Sekundärstruktur S einer RNA-Sequenz A = a1a2…an ist eine Menge von Basenpaaren. Ein Basenpaar zwischen ai und aj ( i < j ) wird notiert als ( i – j ) M = { (i j) | 1 i < j n, (ai aj) ist Basenpaar und jedes i und j taucht max 1 mal auf }
 wird notiert als ( i – j ) M = { (i j) | 1 i < j n, (ai aj) ist Basenpaar und jedes i. und j taucht max 1 mal auf }")
48
Eine Menge von Basenpaaren wird RNA-Sekundärstruktur ohne Pseudoknoten
genannt, wenn folgende Bedingung erfüllt ist: Es existieren keine Basenpaare (ai aj), (ah ak) M, die i h j k erfüllen. Skizze: i…..h…..j…..k
, (ah ak) M, die i h j k. erfüllen. Skizze: i…..h…..j…..k")
49
Eine Menge von Basenpaaren wird RNA-Sekundärstruktur mit Pseudoknoten
genannt, wenn folgende Bedingung erfüllt ist: Es existieren Positionen j‘ und j‘‘ für I < j‘ < j‘‘ < K, so dass für jedes Paar (i j) MI,K gilt: I i < j‘ < j < j‘‘ oder j‘ < i < j‘‘ j K
 MI,K gilt: I i < j‘ < j < j‘‘ oder j‘ < i < j‘‘ j K")
50
Gliederung Einleitung Algorithmus von Akutsu
Nearest Neighbour Thermodynamik Regeln Berechnung minimaler Energien von RNA-Substrukturen Optimale Energie eines Pseudoknots Analyse des Algorithmus

51
Der Algorithmus von Akutsu bewertet RNA-Strukturen anhand der Anzahl
ihrer Basenpaare. Idee: Basenpaarungen tragen zu einer erhöhten Ordnung im Molekül bei und erniedrigen dadurch die freie Energie der Struktur. Strukturen mit hoher Anzahl Basenpaaren werden daher in der Natur bevorzugt und durch den Algorithmus besser bewertet.

52
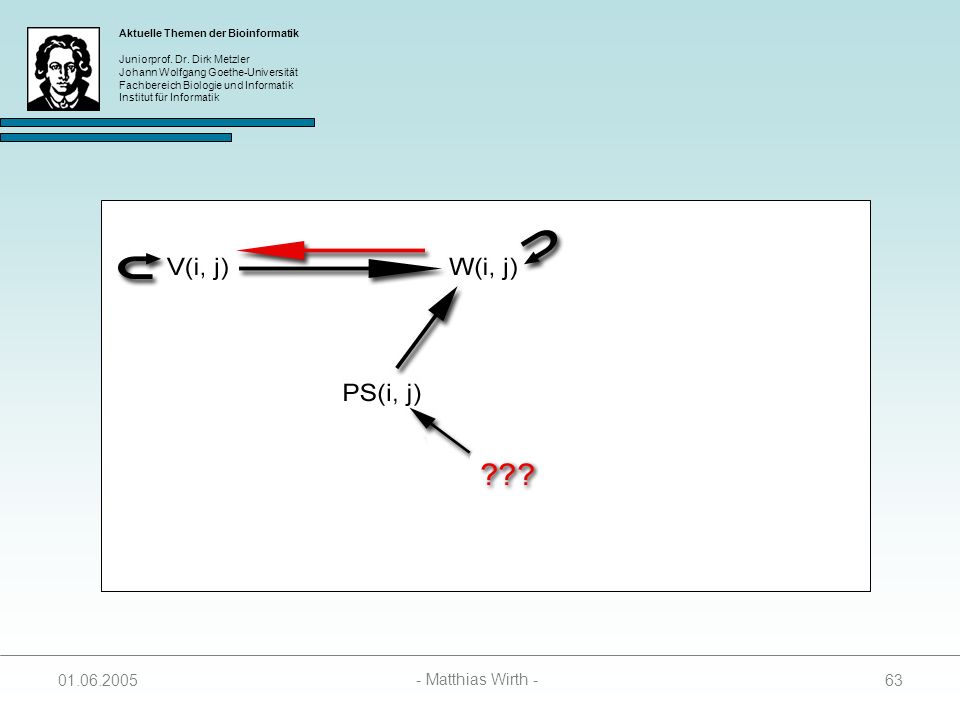
Zur Berechnung des optimalen Pseudoknots werden 4 Matrizen benötigt:
SL(i, j, k) enthält Score des besten Foldings zwischen I und i, und j und k. Unter der Bedingung das i mit j paart. SR(i, j ,k) enthält Score des besten Foldings zwischen I und i, und j und k. Unter der Bedingung das j mit k paart. SM(i, j, k) enthält Score des besten Foldings zwischen I und i, und j und k. Unter der Bedingung das weder i mit j, noch j mit k paart. PS(i, j) enthält Score des besten Pseudoknot mit Anfangspunkt i und Endpunkt j
 enthält Score des besten Foldings zwischen I und i, und. j und k. Unter der Bedingung das i mit j paart. SR(i, j ,k) enthält Score des besten Foldings zwischen I und i, und. j und k. Unter der Bedingung das j mit k paart. SM(i, j, k) enthält Score des besten Foldings zwischen I und i, und. j und k. Unter der Bedingung das weder i mit j, noch j mit. k paart. PS(i, j) enthält Score des besten Pseudoknot mit Anfangspunkt i. und Endpunkt j")
53
Um einen Pseudoknot mit Anfangspunkt I und Endpunkt K zu finden, muss
der Algorithmus drei Typen von Triplets berechnen: SL(i, j, k), SR(i, j, k) und SM(i, j, k) für jedes i, j, k für das gilt (I i < j < k K) Berechnung von SL(i, j, k):
, SR(i, j, k) und SM(i, j, k) für jedes i, j, k für das gilt (I i < j < k K) Berechnung von SL(i, j, k):")
54
Berechnung von SR(i, j, k):
Berechnung von SM(i, j, k):
:")
55
Optimaler Score für jedes Paar (i, j) kann durch folgende Rekursion
Für jedes Paar (I, K), wobei I < K, werden die SL, SM und SR Matrizen berechnet. Optimaler Score für jedes Paar (i, j) kann durch folgende Rekursion berechnet werden:
, wobei I < K, werden die SL, SM und SR Matrizen. berechnet. Optimaler Score für jedes Paar (i, j) kann durch folgende Rekursion. berechnet werden:")
56
Gliederung Einleitung Algorithmus von Akutsu
Nearest Neighbour Thermodynamik Regeln Berechnung minimaler Energien von RNA-Substrukturen Optimale Energie eines Pseudoknots Analyse des Algorithmus

57
Die Nearest Neighbour Energy Rules sind weit verbreitet in der RNA
Sekundärstrukturvorhersage. Problem ist so definiert: Berechnung von RNA-Strukturen mit minimaler freier Energie (-G)
")
58
Es existiert keine systematische Studie über die Thermodynamik
Problem: Es existiert keine systematische Studie über die Thermodynamik von Pseudoknots. In den Nearest Neighbour Energy Rules sind Pseudoknots „verboten“. Annahme: Freie Energie eines Pseudoknots ist die Summe der stabilisierenden Werte beider Stämme und die der destabilisierenden Loops.

59
Gliederung Einleitung Algorithmus von Akutsu
Nearest Neighbour Thermodynamik Regeln Berechnung minimaler Energien von RNA-Substrukturen Optimale Energie eines Pseudoknots Analyse des Algorithmus

60
Zur Berechnung der minimalen Energie von RNA-Substrukturen werden
3 N x N Matrizen benötigt: 1. V(i,j) enthält Score des besten Foldings zwischen i und j, unter der Bedingung das i und j paart. 2. W(i,j) enthält Score des besten Foldings zwischen i und j, egal ob i und j paaren oder nicht. 3. PS(i,j) enthält Score der besten Pseudoknot-Konfiguration zwischen den Positionen i und j. Für jedes RNA-Segment mit Start i und Ende j (i<j)
 enthält Score des besten Foldings zwischen i und j, unter. der Bedingung das i und j paart. 2. W(i,j) enthält Score des besten Foldings zwischen i und j, egal. ob i und j paaren oder nicht. 3. PS(i,j) enthält Score der besten Pseudoknot-Konfiguration zwischen. den Positionen i und j. Für jedes RNA-Segment mit Start i und Ende j (i<j)")
61
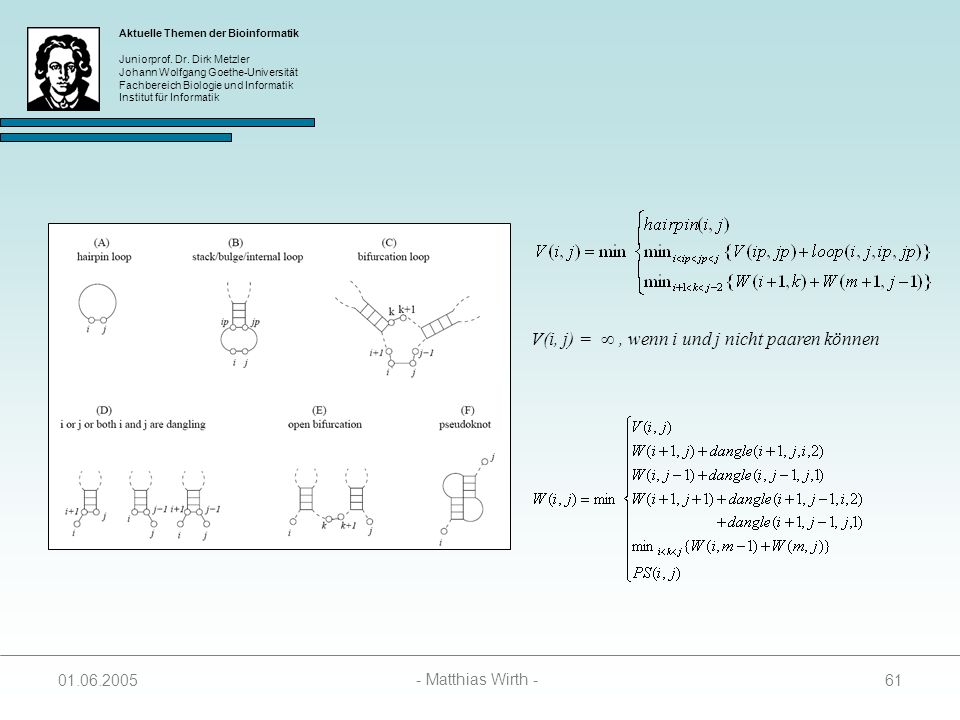
V(i, j) = , wenn i und j nicht paaren können
62
Struktur in jedem Schritt ist.
Algorithmus nimmt immer ein Nukleotid dazu und beobachtet, was die beste Struktur in jedem Schritt ist. Im letzten Schritt wird W(1, n) berechnet und enthält die minimale Energie der gesamten Sequenz. Über ein Traceback durch die Matrizen werden die Strukturen der Sequenz bestimmt.
 berechnet und enthält die minimale Energie. der gesamten Sequenz. Über ein Traceback durch die Matrizen werden die Strukturen der Sequenz. bestimmt")
64
Gliederung Einleitung Algorithmus von Akutsu
Nearest Neighbour Thermodynamik Regeln Berechnung minimaler Energien von RNA-Substrukturen Optimale Energie eines Pseudoknots Analyse des Algorithmus

65
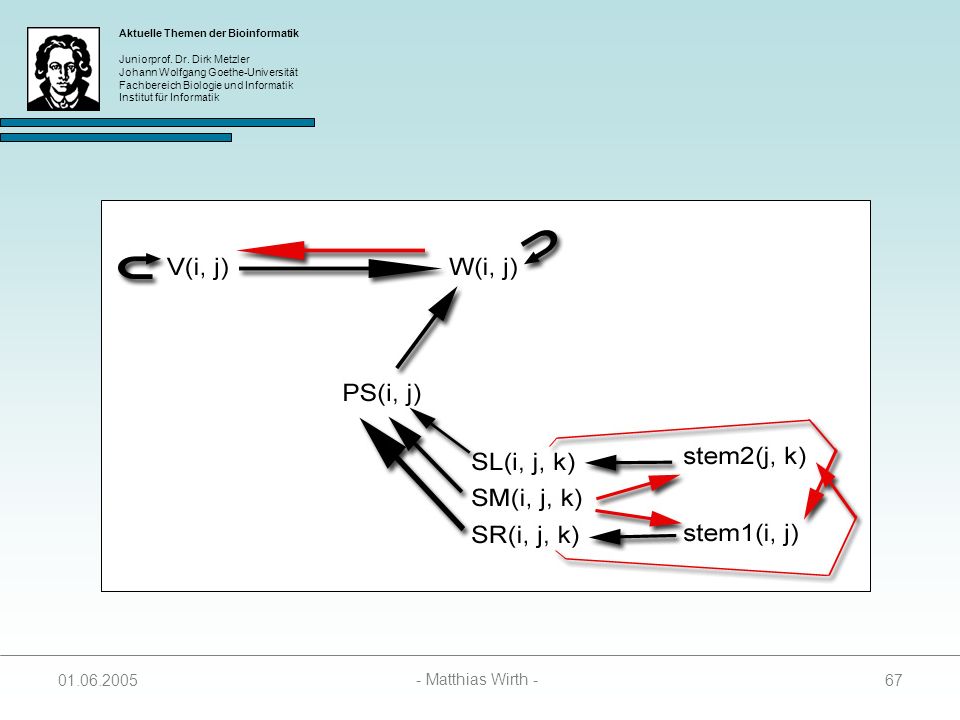
Optimale Energie eines Pseudoknots
Folgende Matrizen werden zur Berechnung benötigt: 1. SL(i, j, k) Enthält Score des besten Folding zwischen Positionen I und i, und j und k. Enthält Energie des Loops der von i und j geschlossen wird. Setzt Paarung von i und j vorraus. 2. SR(i, j, k) Enthält Score des besten Folding zwischen Positionen I und i, und j und k. Enthält Energie des Loops der von i und j+1 geschlossen wird. Setzt Paarung von j und k vorraus. 3. SM(i, j, k) Enthält Score des besten Folding zwischen Positionen I und i, geschlossen wird. Setzt vorraus, dass weder i mit j paart, noch j mit k.
 Enthält Score des besten Folding zwischen Positionen I und i, und j und k. Enthält Energie des Loops der von i und j. geschlossen wird. Setzt Paarung von i und j vorraus. 2. SR(i, j, k) Enthält Score des besten Folding zwischen Positionen I und i, und j und k. Enthält Energie des Loops der von i und j+1. geschlossen wird. Setzt Paarung von j und k vorraus. 3. SM(i, j, k) Enthält Score des besten Folding zwischen Positionen I und i, geschlossen wird. Setzt vorraus, dass weder i mit j paart, noch j mit k")
66
i mit j paart und in SM(i, j, k) falls i nicht mit j paart.
4. stem1(i, j) Enthält Energie von S1, die in SL(i, j, k) gespeichert ist, falls i mit j paart und in SM(i, j, k) falls i nicht mit j paart. 5. stem2(j, k) Enthält Energie von S2, die in SR(i, j, k) gespeichert ist, falls j mit k paart und in SM(i, j, k) falls j nicht mit k paart. stem1 und stem2 werden zur Berechnung von SL, SR und SM benötigt. stem1 und stem2 erhalten die Werte, die als minimale Energien für SL, SR oder SM gewählt wurden. stem1 und stem2 enthalten zusammen die Energie einer Struktur (i, j, k).
 Enthält Energie von S1, die in SL(i, j, k) gespeichert ist, falls. i mit j paart und in SM(i, j, k) falls i nicht mit j paart. 5. stem2(j, k) Enthält Energie von S2, die in SR(i, j, k) gespeichert ist, falls. j mit k paart und in SM(i, j, k) falls j nicht mit k paart. stem1 und stem2 werden zur Berechnung von SL, SR und SM benötigt. stem1 und stem2 erhalten die Werte, die als minimale Energien für SL, SR oder SM gewählt wurden. stem1 und stem2 enthalten zusammen die Energie einer Struktur (i, j, k)")
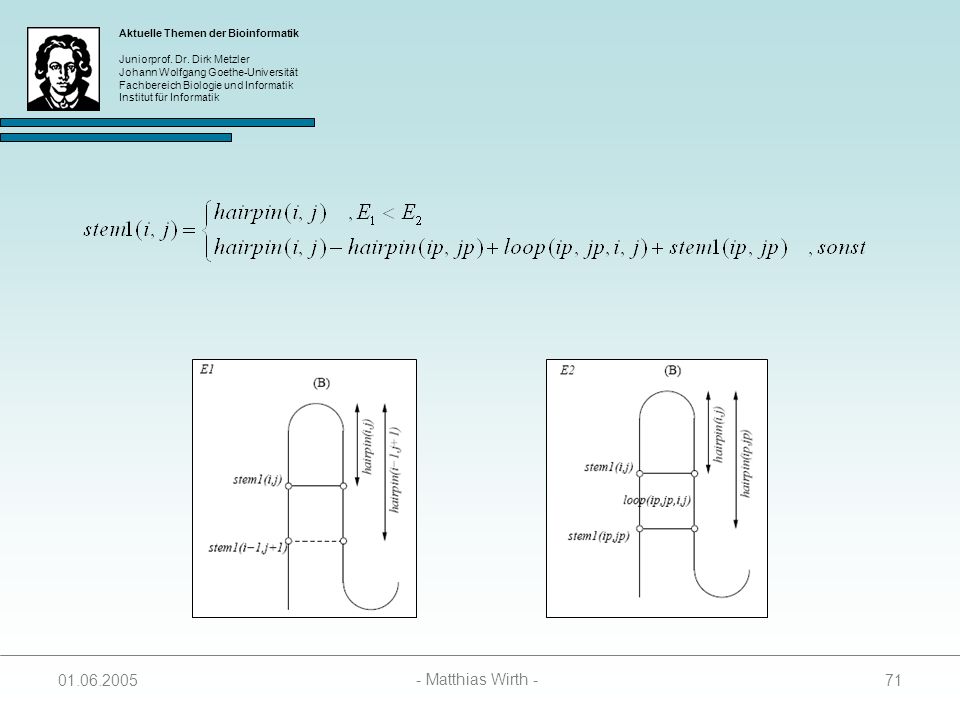
68
SL(i, j, k) = SR(i, j, k) = SM(i, j, k) = für alle i, j, k außer:
Initialisierung SL(i, j, k) = SR(i, j, k) = SM(i, j, k) = für alle i, j, k außer: SL(i, k-1, k) = hairpin(i, k-1) + penalty wenn i und k-1 paaren können stem1(i, j) = hairpin(i, j) falls i und j paaren können stem1(i, j) = sonst stem2(i, j) = für alle i, j
 = SR(i, j, k) = SM(i, j, k) = für alle i, j, k. außer: SL(i, k-1, k) = hairpin(i, k-1) + penalty wenn i und k-1 paaren können. stem1(i, j) = hairpin(i, j) falls i und j paaren können. stem1(i, j) = sonst. stem2(i, j) = für alle i, j")
69
Berechnung der SL Matrix
Wenn i und j paaren, kann der Wert in SL(i, j, k) auf drei Arten zustandekommen: Das Paar (i – j) schließt einen Hairpin Loop 2. Das Paar (i – j) stackt auf einem Paar (i-1 – j+1) 3. Das Paar (i – j) schließt zusammen mit einem Paar (ip – jp) einen Bulge oder einen Internal Loop
 auf drei Arten. zustandekommen: Das Paar (i – j) schließt einen Hairpin Loop. 2. Das Paar (i – j) stackt auf einem Paar (i-1 – j+1) 3. Das Paar (i – j) schließt zusammen mit einem Paar (ip – jp) einen Bulge oder einen Internal Loop")
70
SL(i, j, k) = min { E1, E2 } E1 = hairpin(i, j) + stem2(j+1, k)
E2 = minIi, i+4 j<jp<k { hairpin(i,j) – hairpin(ip, jp) + loop(ip, jp, i, j) + SL(ip, jp, k) }
 – hairpin(ip, jp) + loop(ip, jp, i, j) + SL(ip, jp, k) }")
72
E1 = hairpin(i,j) + penalty
Spezialfall: Ist stem2(j+1, k) = ( Substruktur enthält nur einen Hairpin-Loop), dann folgt: E1 = hairpin(i,j) + penalty Paaren i und j nicht, werden SL(i, j, k) und stem1(i, j) wie folgt berechnet:
 = ( Substruktur enthält nur einen Hairpin-Loop), dann folgt: E1 = hairpin(i,j) + penalty. Paaren i und j nicht, werden SL(i, j, k) und stem1(i, j) wie folgt berechnet:")
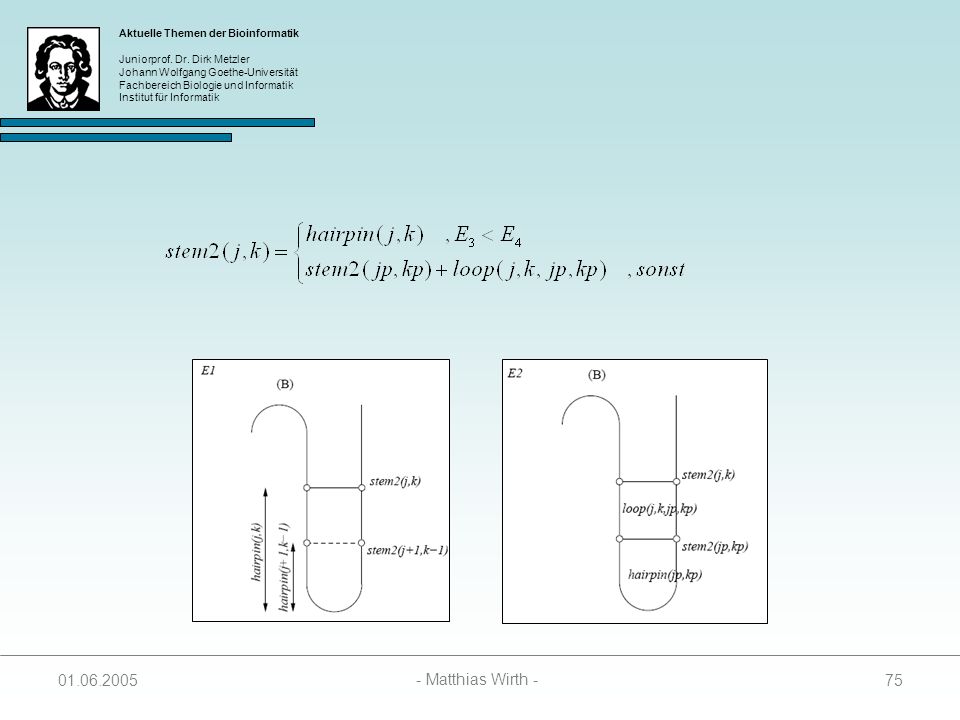
73
Berechnung der SR Matrix
Wenn j und k paaren, kann der Wert in SL(i, j, k) auf drei Arten zustandekommen: Das Paar (j – k) schließt einen Hairpin Loop 2. Das Paar (j – k) stackt auf einem Paar (j+1 – k-1) 3. Das Paar (j – k) schließt zusammen mit einem Paar (jp – kp) einen Bulge oder einen Internal Loop
 auf drei Arten. zustandekommen: Das Paar (j – k) schließt einen Hairpin Loop. 2. Das Paar (j – k) stackt auf einem Paar (j+1 – k-1) 3. Das Paar (j – k) schließt zusammen mit einem Paar (jp – kp) einen Bulge oder einen Internal Loop")
74
SR(i, j, k) = min { E3, E4 } E3 = hairpin(j, k) + stem1(i, j+1)
E4 = minj<jp, jp+4kp<k { loop(j, k, jp, kp) + SL(i, jp, kp) }
 + SL(i, jp, kp) }")
76
E3 = hairpin(j, k) + penalty
Spezialfall: Ist stem1(i, j+1) = ( Substruktur enthält nur einen Hairpin-Loop), dann folgt: E3 = hairpin(j, k) + penalty Paaren j und k nicht, werden SR(i, j, k) und stem2(j, k) wie folgt berechnet:
 = ( Substruktur enthält nur einen Hairpin-Loop), dann folgt: E3 = hairpin(j, k) + penalty. Paaren j und k nicht, werden SR(i, j, k) und stem2(j, k) wie folgt berechnet:")
77
Berechnung der SM Matrix
In der SM Matrix geht man davon aus, dass weder i mit j, noch j mit k paaren, auch wenn sie dazu in der Lage wären. Bei Fall 1.) stem1(i, j) = stem1(i-1, j) Bei Fall 2.) stem1(i, j) = stem1(i, j+1), stem2(j, k) = stem2(j+1, k) Bei Fall 3.) stem2(j, k)= stem2(j, k-1)
 stem1(i, j) = stem1(i-1, j) Bei Fall 2.) stem1(i, j) = stem1(i, j+1), stem2(j, k) = stem2(j+1, k) Bei Fall 3.) stem2(j, k)= stem2(j, k-1)")
78
Der Score eines Triplets hängt nur von I ab nicht von K.
Komplexität: Für jedes Paar (I, K) müssen Scores für (n³) Triplets berechnet werden. Der Score eines Triplets hängt nur von I ab nicht von K. Es müssen (n³) Scores für jedes I berechnet werden Zeit: (n4) Der Speicherplatzbedarf resultiert aus den NxNxN-Matrizen Speicherplatz: (n³)
 müssen Scores für (n³) Triplets berechnet werden. Der Score eines Triplets hängt nur von I ab nicht von K. Es müssen (n³) Scores für jedes I berechnet werden. Zeit: (n4) Der Speicherplatzbedarf resultiert aus den NxNxN-Matrizen. Speicherplatz: (n³)")
79
Gliederung Einleitung Algorithmus von Akutsu
Nearest Neighbour Thermodynamik Regeln Berechnung minimaler Energien von RNA-Substrukturen Optimale Energie eines Pseudoknots Analyse des Algorithmus

80
Test mit einer Menge von simplen Pseudoknots aus PseudoBase
Ergebnisse: Test mit einer Menge von simplen Pseudoknots aus PseudoBase 169 Sequenzen, mit einer Länge zwischen 19 und 114 Nukleotiden Algorithmus faltet 163 Pseudoknots und 6 einfache Strukturen 131/163 sind korrekt oder fast korrekt gefaltet worden Für 3 der 6 einfachen Strukturen kann die Vorhersage, durch Erhöhen der penalty verbessert werden Bei einer der simplen Strukturen ist im Pseudoknot der Datenbank ein A-G bp enthalten

81
Vergleich mit dem Eddy/Rivas Programm:
Deogun/Komina 50 % der Pseudoknots erkannt 95 % der Pseudoknots erkannt Davon 78 % mit korrekter oder fast-korrekter Struktur Berechnungszeiten: 75 Nukleotide 55 Sekunden 114 Nukleotide 8 Minuten

82
Quellen: Akutsu (2000): Dynamic programming algorithm for RNA secondary structure prediction with pseudoknots, Discrete Apllied Mathematics Deogun, Komina et al. (2004): RNA Secondary Structure Prediction with Simple Pseudoknots, APBC2004 Holmes (2004): A probabilistic model for the evolution of RNA structure, BMC Bioinformatics Mattick (2005): Das verkannte Genom-Programm, Spektrum der Wissenschaft (März 05) Thorne, Kishino, Felsenstein (1991): An evolutionary model for maximum likelihood alignment of DNA sequences, J Mol Evol Zuker et al.: Algorithms and thermodynamics for RNA secondary structure prediction: A practical guide, NATO ASI Series
: RNA Secondary Structure Prediction with Simple Pseudoknots, APBC2004. Holmes (2004): A probabilistic model for the evolution of RNA structure, BMC Bioinformatics. Mattick (2005): Das verkannte Genom-Programm, Spektrum der Wissenschaft (März 05) Thorne, Kishino, Felsenstein (1991): An evolutionary model for maximum likelihood alignment of DNA sequences, J Mol Evol. Zuker et al.: Algorithms and thermodynamics for RNA secondary structure prediction: A practical guide, NATO ASI Series")
Ähnliche Präsentationen




![Definition [1]: Sei S eine endliche Menge und sei p eine Abbildung von S in die positiven reellen Zahlen Für einen Teilmenge ES von S sei p definiert.](/1/209241/big_thumb.jpg "Definition [1]: Sei S eine endliche Menge und sei p eine Abbildung von S in die positiven reellen Zahlen Für einen Teilmenge ES von S sei p definiert.>")


>")









 Matrixkettenprodukt>")
 Prof. Th. Ottmann.>")
 Matrixkettenprodukt Prof. Dr. Th. Ottmann.>")
 Prof. Th. Ottmann.>")
 T. Lauer.>")