Präsentation herunterladen
Die Präsentation wird geladen. Bitte warten

1
Vorlesung Bioinformatik Teil II
Genomics 04.06.: Genomstrukturen, Sequenzierprojekte 11.06.: Annotation, Datenbanken und Datenbanksuche 18.06.: Paarweiser Sequenzvergleich (Rainer Merkl) 25.06.: Multipler Sequenzvergleich, Anwendungen (Rainer Merkl)
 : Multipler Sequenzvergleich, Anwendungen (Rainer Merkl)")
2
Genom-Sequenzierung und Auswertung der Daten
Genome und Gene Genom-Sequenzierung und Auswertung der Daten

3
Genomstrukturen Unterschiede zwischen Pro- und Eukaryonten:
Prokaryonten haben keinen Zellkern und keine Organelle Eukaryonten können bis zu zwei, vom Kern unabhängige, Genome aufweisen Mitochondrien: 16 bis 100 kb; bei Pflanzen bis kb Chloroplasten: 100 bis 300 kb

4
Genomgrößen und Genanzahl

5
Evolution der Genomgröße

6
Bakterien-Genome Genom ist meist eine ringförmige DNA
im Nucleoid lokalisiert viele, manchmal überlappende Gene z. B. E. coli: ca. 1 Gen pro kb Gene selbst sind nicht unterbrochen, ca. 1 kb groß Operons: mehrere Gene hinter einem Promotor Haushaltsgene bis zu Säugern konserviert

7
Eukaryonten-Genome Genom ist im Kern lokalisiert
meist in mehreren Chromosomen Gene oft weit getrennt z. B. H. sapiens: ca. 9 Gene pro Mb, ca kb groß Gene selbst sind oft unterbrochen (Introns) viele Bereiche nicht-kodierend
 viele Bereiche nicht-kodierend.")
8
Eukaryonten-Genstruktur
primäres Transkript fertige mRNA Unterschiede beim Prokaryonten (Bakterien): - andere Promotorstruktur - UTRs sehr kurz - keine Introns - andere Termination (kein polyA)
: - andere Promotorstruktur - UTRs sehr kurz - keine Introns - andere Termination (kein polyA)")
9
Eukaryonten-Genome

10
30.000 Gene beim Menschen Größe der Gene: ca. 10 - 15 kb
Abstand zwischen Genen: ca kb "Rekordhalter": Dystrophin-Gen mit über 2,4 Mb (79 Exons) Größe der Exons: ca. 170 bp, aber auch Ausnahmen bis zu 7,6 kb Anzahl der Introns: 0 (z. B. Histon H4) bis 118 (Typ7-Collagen, 31 kb) Größe der Introns: bis hin zu 150 kb Größe der mRNAs: ca. 2,2 kb mit großer Variabilität Gen ist nicht gleich Gen: Benutzung alternativer Promotoren (z. B. 7 beim Dystrophin-Gen) alternatives Spleißen alternative Polyadenylierung
 Größe der Exons: ca. 170 bp, aber auch Ausnahmen bis zu 7,6 kb. Anzahl der Introns: 0 (z. B. Histon H4) bis 118 (Typ7-Collagen, 31 kb) Größe der Introns: bis hin zu 150 kb. Größe der mRNAs: ca. 2,2 kb mit großer Variabilität. Gen ist nicht gleich Gen: Benutzung alternativer Promotoren (z. B. 7 beim Dystrophin-Gen) alternatives Spleißen. alternative Polyadenylierung.")
11
Von der Karte zum Genom Chromosomenkarte des X-Chromosoms

12
Von der Karte zum Genom

13
Strategien zur Genomsequenzierung
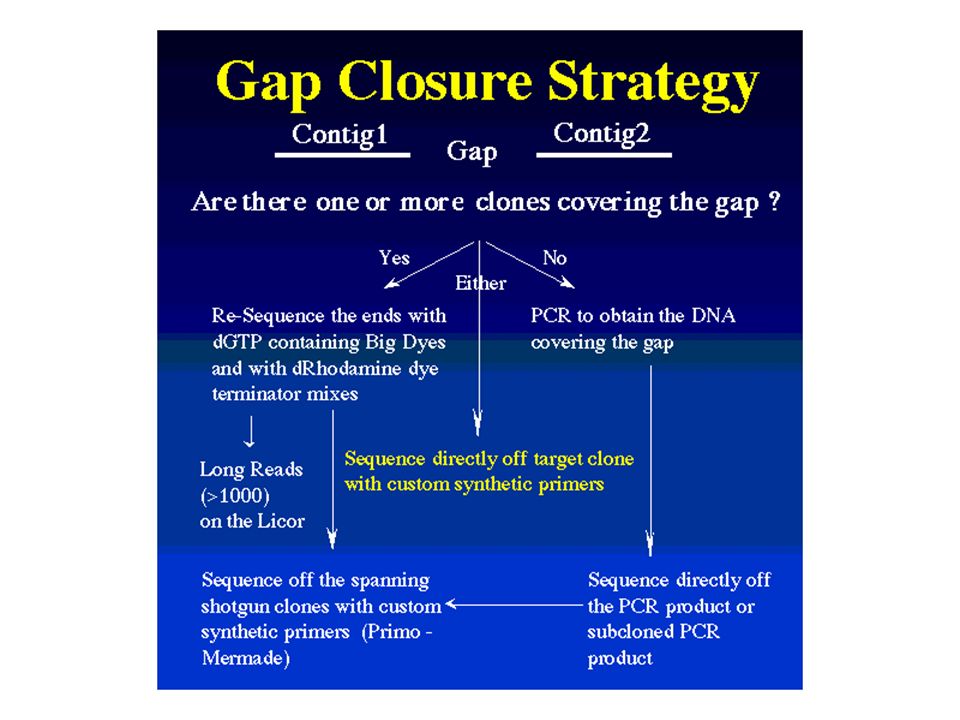
ESTs (expressed sequence tags) für exprimierte Gene (cDNA) Shot gun- oder Primer Walk-Methode (genomisch) Klonierung in Cosmide, BACs (Bacterial Artificial Chromosomes), PACs (P1-derived artificial chromosome) Phagemide, P1-Phagen Insert-Größenverteilung in BACs
 für exprimierte Gene (cDNA) Shot gun- oder Primer Walk-Methode (genomisch) Klonierung in Cosmide, BACs (Bacterial Artificial Chromosomes), PACs (P1-derived artificial chromosome) Phagemide, P1-Phagen. Insert-Größenverteilung in BACs.")
14
Shot gun - Sequenzierung
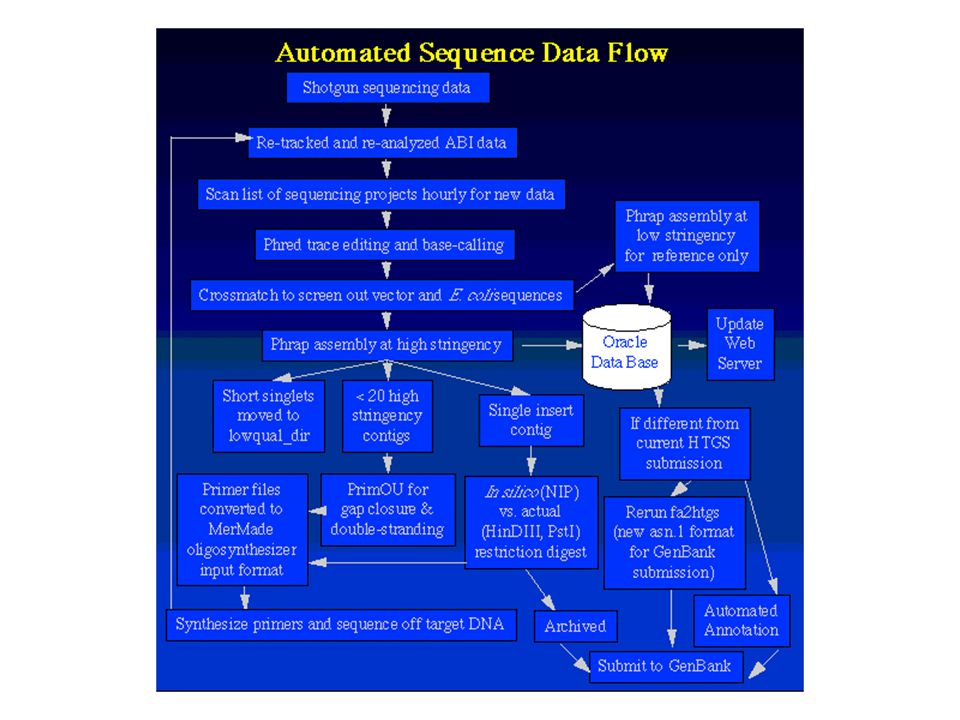
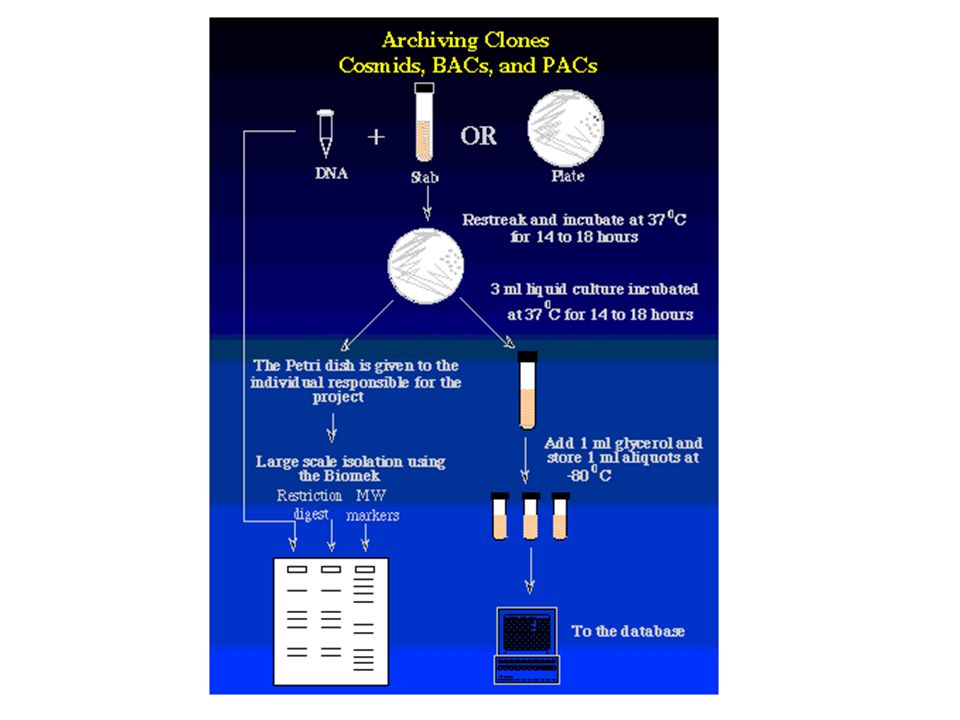
Zufällig fragmentierte DNA durch "Nebulizer" Klonierung in Standard-Vektoren High-throughput-Sequenzierung durch automatische Sequencer (z. B. ABI377) und Fluoreszenz-markierte Terminatoren (ddNTPs); Standard-Primer Zusammenfügen der Sequenzstücke durch Assembly-Programme Untersuchung auf biologisch relevante DNA-Sequenzen durch Datenbankabfragen Klone werden nach Analyse aufbewahrt
 und Fluoreszenz-markierte Terminatoren (ddNTPs); Standard-Primer. Zusammenfügen der Sequenzstücke durch Assembly-Programme. Untersuchung auf biologisch relevante DNA-Sequenzen durch Datenbankabfragen. Klone werden nach Analyse aufbewahrt.")
20
Das "Assembly"-Problem Das Ergebnis einer Shot gun - Sequenzierung ist vergleichbar mit einem Puzzle: - Viele, viele Teile - Vorder- und Rückseite - Einige sind schmutzig oder unkenntlich - Teile anderer Puzzle sind mit drin Multiplizität der Probleme: - 99% Lesegenauigkeit bei der Sequenzierung, d. h. Ø 1 Fehler pro 100 bp - Häufung der Fehler am Ende der Sequenz - genau diese Regionen sind für das Assembly wichtig - repetitive Sequenzen in der DNA, gleiche Sequenzen können auf verschiedenen Chromosomen auftreten Assembly der Sequenzen ist der Flaschenhals der Genomprojekte

21
Das "Assembly"-Problem Fehlertolerante Algorithmen zum Alignment zweier und mehrerer Sequenzen Fehlerquellen: - primäre Fehler: chemisch, d. h. bei der DNA-Gewinnung (v. a. PCR) oder bei der Sequenzreaktion - sekundäre Fehler: beim Lesen des Chromatogramms (suboptimale Signalqualität; Lösung: menschliche Erfahrung und bessere Chemie, v. a. Dyes) - tertiäre Fehler: Klonierungsvektorsequenzen müssen entfernt werden Probleme: Effizienz und Automatisierung Effizienz: Das Ausgabeformat (SCF: Standard Chromatography Format) der Sequenzer benötigt relativ viel Speicherkapazität (ca. 100 byte pro Base, d. h Reaktionen mit je gelesenen Basen benötigen 100 MB) geeigneteres Format (CAF: Common Assembly Format, standardisiert) Automatisierung: Kombination des Alignment-Reject-Editing-Verfahrens in silicio
 oder bei der Sequenzreaktion - sekundäre Fehler: beim Lesen des Chromatogramms (suboptimale Signalqualität; Lösung: menschliche Erfahrung und bessere Chemie, v. a. Dyes) - tertiäre Fehler: Klonierungsvektorsequenzen müssen entfernt werden. Probleme: Effizienz und Automatisierung. Effizienz: Das Ausgabeformat (SCF: Standard Chromatography Format) der Sequenzer benötigt relativ viel Speicherkapazität (ca. 100 byte pro Base, d. h Reaktionen mit je gelesenen Basen benötigen 100 MB) geeigneteres Format (CAF: Common Assembly Format, standardisiert) Automatisierung: Kombination des Alignment-Reject-Editing-Verfahrens in silicio.")
22
Das "Assembly"-Problem Alignment: Alphabet mit allen Zeichen, die im Alignment vorkommen (können): = {A,C,G,T,*,~} Die Sequenz S ist eine geordnete Folge von Charakteren aus dem Alphabet : S = {s1,...,sn} mit n = |S| und si Durch die Einführung von "end-gaps" (~) in das Alignment wird das Problem umgangen, daß alle Sequenzen die gleiche Länge haben müssen. Bsp.: Sequenz 1: ACGTACGTACGTACGTACGTACGT~~~~ Sequenz 2: ~~~~~CG*ACGT*CGTACGTACGTACGT
 in das Alignment wird das Problem umgangen, daß alle Sequenzen die gleiche Länge haben müssen. Bsp.: Sequenz 1: ACGTACGTACGTACGTACGTACGT~~~~ Sequenz 2: ~~~~~CG*ACGT*CGTACGTACGTACGT.")
23
Das "Assembly"-Problem Bewertung des Alignments: Der numerische Vergleich zweier Elemente in einem Alignment wird als Score bezeichnet: score(s1, s2) Der Score-Wert einer Spalte in einem Alignment ist die Summe der Scores der Permutation von Elementen dieser Spalte: k k score(s1,...,sk) = score(sj,sm) j=1 m=j Der Score-Wert des Gesamt-Alignments ist demnach die Summe aller Spalten-Scores: n k k score(S1,...,Sk) = score(sj,sm) i j m
 Der Score-Wert einer Spalte in einem Alignment ist die Summe der Scores der Permutation von Elementen dieser Spalte: k k score(s1,...,sk) = score(sj,sm) j=1 m=j. Der Score-Wert des Gesamt-Alignments ist demnach die Summe aller Spalten-Scores: n k k score(S1,...,Sk) = score(sj,sm) i j m.")
24
Das "Assembly"-Problem Accept/Reject des Alignments: Für jede Art von Abweichungen können "Strafpunkte" vergeben werden: - Direkte Abweichung: „mismatch“ (z. B. T-A, C-T etc.) - Einfügen von Lücken - Verlängerung von Lücken - Lücken am Ende Erreichen die Strafpunkte einen Schwellenwert (threshold), so wird das Alignment abgelehnt; ansonsten wird das Ergebnis gespeichert und mit anderen Alignments weiter verglichen.
 - Einfügen von Lücken - Verlängerung von Lücken - Lücken am Ende Erreichen die Strafpunkte einen Schwellenwert (threshold), so wird das Alignment abgelehnt; ansonsten wird das Ergebnis gespeichert und mit anderen Alignments weiter verglichen.")
25
Einfachster Algorithmus für Alignments: Dotplot
Gegeben: A = a1,a2,a3,...,an Sequenz A der Länge n B = b1,b2,b3,...,bm Sequenz B der Länge m A C G T Für alle i, j mit 1 i n, 1 j m soll gelten: M [i,j] = 1 für ai = bj score für match M [i,j] = 0 für aj bj score für mismatch M [i,j] wird als 2-dimensionale Matrix dargestellt

26
Einfachster Algorithmus für Alignments: Dotplot
Gegeben: A = a1,a2,a3,...,an Sequenz A der Länge n B = b1,b2,b3,...,bm Sequenz B der Länge m A C G T 1 Für alle i, j mit 1 i n, 1 j m soll gelten: M [i,j] = 1 für ai = bj score für match M [i,j] = 0 für aj bj score für mismatch M [i,j] wird als 2-dimensionale Matrix dargestellt

27
Einfachster Algorithmus für Alignments: Dotplot
Gegeben: A = a1,a2,a3,...,an Sequenz A der Länge n B = b1,b2,b3,...,bm Sequenz B der Länge m A C G T 1 Für alle i, j mit 1 i n, 1 j m soll gelten: M [i,j] = 1 für ai = bj score für match M [i,j] = 0 für aj bj score für mismatch M [i,j] wird als 2-dimensionale Matrix dargestellt Längste Diagonale ohne Unterbrechung Markiert das beste Teilalignment

28
Dotplot-Beispiel: Sequenzassembly
Sequenz B Sequenz B Grafische Darstellungsmöglichkeit: 1 (match) weißer Punkt 0 (mismatch) schwarzer Punkt
 weißer Punkt. 0 (mismatch) schwarzer Punkt.")
29
Accept/Reject des Alignments

30
„Feinheiten“ des Assembly-Problems

31
Dotplot-Beispiel: Vergleich cDNA (mRNA) – genom. DNA
k a t A T Polyadenylierungs- l Stopcodon stelle a n s T r + 1 n TAG AAUAAA Promotor a mRNA (cDNA) T r Exon Intron genom. DNA 5’ UTR 3’ UTR Transkription Spleißen genom. DNA mRNA Cap AAAAA
 T. r. Exon. Intron. genom. DNA. 5’ UTR. 3’ UTR. Transkription. Spleißen. genom. DNA. mRNA. Cap. AAAAA.")
32
Notwendigkeit von Scoring-Matrizen
Nukleotid-Sequenzalignments können über scoring-Kritierien wie scorematch = 1 scoremismatch = 0 bewertet werden. Bei Protein-Sequenzen ist dies nicht mehr realistisch: Ein Austausch einer Aminosäure gegen eine ähnliche ist anders zu bewerten als ein Austausch gegen eine unähnliche.

33
Ähnlichkeitsmatrizen
BLOSUM45 Amino Acid Similarity Matrix (BLOcks SUbstituition Matrix) Gly 7 Pro Asp Glu Asn His Gln Lys Arg Ser Thr Ala Met Val Ile Leu Phe Tyr Trp Cys Gly Pro Asp Glu Asn His Gln Lys Arg Ser Thr Ala Met Val Ile Leu Phe Tyr Trp Cys
 Gly 7. Pro Asp Glu Asn His Gln Lys Arg Ser Thr Ala Met Val Ile Leu Phe Tyr Trp Cys Gly Pro Asp Glu Asn His Gln Lys Arg Ser Thr Ala Met Val Ile Leu Phe Tyr Trp Cys.")
34
Vorlesung Bioinformatik Teil II
Genomics 04.06.: Genomstrukturen, Sequenzierprojekte 11.06.: Annotation, Datenbanken und Datenbanksuche 18.06.: Paarweiser Sequenzvergleich (Rainer Merkl) 25.06.: Multipler Sequenzvergleich, Anwendungen (Rainer Merkl)
 : Multipler Sequenzvergleich, Anwendungen (Rainer Merkl)")
35
Historisches zu Datenbanken
"Zuerst war das Protein" Dayhoff, Anfang der 60er Jahre: Sammlung von allen bekannten Aminosäuresequenzen Atlas of Protein Sequences and Structures (Dayhoff et al., 1965) Grundlage für die PIR-Datenbank (Protein Information Resource) EMBL-Nukleotiddatenbank (1982) erste DNA-Sequenzdatenbank am European Molecular Biology Laboratory in Hinxton, England - mit DDBJ (Mishima, Japan) und NCBI (Bethesda, USA) in der "International Nucleotide Sequence Database Collaboration" (1988) - separate Eingabe möglich, aber täglicher Datenabgleich - Updates nur bei der Stelle möglich, bei der der Record erzeugt wurde
 Grundlage für die PIR-Datenbank (Protein Information Resource) EMBL-Nukleotiddatenbank (1982) erste DNA-Sequenzdatenbank am European Molecular Biology Laboratory in Hinxton, England. - mit DDBJ (Mishima, Japan) und NCBI (Bethesda, USA) in der International Nucleotide Sequence Database Collaboration (1988) - separate Eingabe möglich, aber täglicher Datenabgleich - Updates nur bei der Stelle möglich, bei der der Record erzeugt wurde.")
36
Wachstum der EMBL-Datenbank
Stand : This morning the EMBL Database contained 66,139,788,831 nucleotides in 40,066,073 entries. Quelle:

37
Datenbankformate GenBank: Genetische Sequenz-Datenbank
gepflegt durch das NCBI (National Center for Biotechnology Information) am NIH (National Institutes of Health), Bethesda, Maryland, USA annotierte Sammlung aller öffentlich verfügbarer Nukleotid- und Proteinsequenzen einzelne Datensätze repräsentieren zusammenhängende DNA- oder RNA-Bereiche mit weiteren Daten (die sogenannte Annotation)
 am NIH (National Institutes of Health), Bethesda, Maryland, USA. annotierte Sammlung aller öffentlich verfügbarer Nukleotid- und Proteinsequenzen. einzelne Datensätze repräsentieren zusammenhängende DNA- oder RNA-Bereiche mit weiteren Daten (die sogenannte Annotation)")
38
NCBI's Entrez Nukleotiddaten als "Sprungbrett" für weitere Informationen, vor allem CDS CDS = Coding Sequence(s), also Translationsinformation von Proteinen Co-Management von DNA- und Proteindaten Analoges System am EBI: Sequence Retrieval System (SRS)
, also Translationsinformation von Proteinen. Co-Management von DNA- und Proteindaten. Analoges System am EBI: Sequence Retrieval System (SRS)")
39
Primäre und sekundäre Datenbanken
Primäre Datenbanken: - experimentelle Ergebnisse - mit einigen Interpretationen (s. u.) - aber ohne kritischen "Review“ - normalerweise direkt von den Forschern mit Daten versorgt Annotation: - CDS (meist abgeleitet von DNA-Sequenz, nicht experimentell) - (mögliche) Funktion (meist durch subjektive Interpretation von Ähnlichkeitsanalysen) - regulatorische Elemente - ...
 - aber ohne kritischen Review - normalerweise direkt von den Forschern mit Daten versorgt. Annotation: - CDS (meist abgeleitet von DNA-Sequenz, nicht experimentell) - (mögliche) Funktion (meist durch subjektive Interpretation von Ähnlichkeitsanalysen) - regulatorische Elemente")
40
Primäre und sekundäre Datenbanken
- abgeleitete Eigenschaften als Haupteintrag - Proteindatenbanken PIR, SWISS-PROT, PDB - abgeleitet aus DNA-Datenbanken - oder direkt eingegeben - oder aus Publikationen übernommen - aber immer soweit wie möglich überprüft

41
Format und Inhalt - Datenbankeinträge: Rohdaten und Annotation
- Verarbeitungseffizienz im Computer und die Verständlichkeit stehen im Widerspruch Beispiel: GenBank-Flatfile bzw. EMBL-Record vs. ASN.1-Record

42
GenBank-Flatfile: . LOCUS LISOD 756 bp DNA BCT 30-JUN-1993
DEFINITION L.ivanovii sod gene for superoxide dismutase. ACCESSION X64011 S78972 NID g44010 VERSION X GI:44010 KEYWORDS sod gene; superoxide dismutase. SOURCE Listeria ivanovii. ORGANISM Listeria ivanovii Bacteria; Firmicutes; Bacillus/Clostridium group; Bacillaceae; Listeria. REFERENCE 1 (bases 1 to 756) AUTHORS Haas,A. and Goebel,W. TITLE Cloning of a superoxide dismutase gene from Listeria ivanovii by functional complementation in Escherichia coli and characterization of the gene product JOURNAL Mol. Gen. Genet. 231 (2), (1992) MEDLINE FEATURES Location/Qualifiers source /organism="Listeria ivanovii" /strain="ATCC 19119" /db_xref="taxon:1638" RBS /gene="sod" gene .
 AUTHORS Haas,A. and Goebel,W. TITLE Cloning of a superoxide dismutase gene from Listeria ivanovii by. functional complementation in Escherichia coli and characterization. of the gene product. JOURNAL Mol. Gen. Genet. 231 (2), (1992) MEDLINE FEATURES Location/Qualifiers. source /organism= Listeria ivanovii /strain= ATCC /db_xref= taxon:1638 RBS /gene= sod gene")
43
Aufbau des GenBank-Flatfiles
1. Header: Informationen, die den gesamten Eintrag betreffen - LOCUS (einmalige accession number, z. B. AF / Länge / Molekülart / Klassifizierung / Datum der letzten Änderung) - DEFINITION (Information, die u. a. bei BLAST mitausgegeben wird) - ACCESSION (primäre und sekundäre accession numbers) - NID (gi number: GenInfo Identifier, wird bei update erneuert) - VERSION (updates) - KEYWORDS (Schlüsselwörter; "historischer Ballast") - SOURCE (gebräuchlicher Name des Organismus, z. B. fruit fly) - ORGANISM (lateinischer Name der Art, z. B. Drosophila melanogaster) - REFERENCE (Publikation, soweit vorhanden, und GenBank-Submission)
 - DEFINITION (Information, die u. a. bei BLAST mitausgegeben wird) - ACCESSION (primäre und sekundäre accession numbers) - NID (gi number: GenInfo Identifier, wird bei update erneuert) - VERSION (updates) - KEYWORDS (Schlüsselwörter; historischer Ballast ) - SOURCE (gebräuchlicher Name des Organismus, z. B. fruit fly) - ORGANISM (lateinischer Name der Art, z. B. Drosophila melanogaster) - REFERENCE (Publikation, soweit vorhanden, und GenBank-Submission)")
44
Aufbau des GenBank-Flatfiles
2. Feature Table: Eigenschaften der Sequenz (FEATURES) - biologische Information - Annotation - z. B. SOURCE / CDS - genaue Übersicht über alle möglichen Einträge in die Feature Table:
 - biologische Information. - Annotation. - z. B. SOURCE / CDS. - genaue Übersicht über alle möglichen Einträge in die Feature Table:")
45
FEATURES Location/Qualifiers
source /organism="Mus musculus" /strain="CD1" promoter <1..9 /gene="ubc42" mRNA join( , ) CDS join( , ) /product="ubiquitin conjugating enzyme" /function="cell division control" /translation="MVSSFLLAEYKNLIVNPSEHFKISVNEDNLTEGPPDTLY QKIDTVLLSVISLLNEPNPDSPANVDAAKSYRKYLYKEDLESYPMEKSLDECS AEDIEYFKNVPVNVLPVPSDDYEDEEMEDGTYILTYDDEDEEEDEEMDDE" exon /number=1 intron exon /number=2 polyA_signal
 CDS join( , ) /product= ubiquitin conjugating enzyme /function= cell division control /translation= MVSSFLLAEYKNLIVNPSEHFKISVNEDNLTEGPPDTLY. QKIDTVLLSVISLLNEPNPDSPANVDAAKSYRKYLYKEDLESYPMEKSLDECS. AEDIEYFKNVPVNVLPVPSDDYEDEEMEDGTYILTYDDEDEEEDEEMDDE exon /number=1. intron exon /number=2. polyA_signal")
46
Aufbau des GenBank-Flatfiles
3. Sequenz: - Formatierte DNA-Sequenz (10er Blöcke zur Übersichtlichkeit) - mit Basenzählung - durchnummeriert - Bsp.: BASE COUNT 1510 a 1074 c 835 g 1609 t 1 gatcctccat atacaacggt atctccacct caggtttaga tctcaacaac ggaaccattg 61 ccgacatgag acagttaggt atcgtcgaga gttacaagct aaaacgagca gtagtcagct 121 ctgcatctga agccgctgaa gttctactaa gggtggataa catcatccgt gcaagaccaa .
 - mit Basenzählung. - durchnummeriert. - Bsp.: BASE COUNT 1510 a 1074 c 835 g 1609 t. 1 gatcctccat atacaacggt atctccacct caggtttaga tctcaacaac ggaaccattg. 61 ccgacatgag acagttaggt atcgtcgaga gttacaagct aaaacgagca gtagtcagct. 121 ctgcatctga agccgctgaa gttctactaa gggtggataa catcatccgt gcaagaccaa. .")
47
EMBL-Record: . . . ID LISOD standard; DNA; PRO; 756 BP. XX
AC X64011; S78972; DT 28-APR-1992 (Rel. 31, Created) DE L.ivanovii sod gene for superoxide dismutase KW sod gene; superoxide dismutase. OS Listeria ivanovii OC Bacteria; Firmicutes; Bacillus/Clostridium group; OC Bacillus/Staphylococcus group; Listeria. RN [1] RX MEDLINE; RA Haas A., Goebel W.; RT "Cloning of a superoxide dismutase gene"; RL Mol. Gen. Genet. 231: (1992). FH Key Location/Qualifiers FH FT source FT /db_xref="taxon:1638" FT /organism="Listeria ivanovii" FT /strain="ATCC 19119" FT RBS FT /gene="sod" FT terminator FT CDS FT /db_xref="SWISS-PROT:P28763" FT /transl_table=11 EMBL-Record: . . .
 DE L.ivanovii sod gene for superoxide dismutase. KW sod gene; superoxide dismutase. OS Listeria ivanovii. OC Bacteria; Firmicutes; Bacillus/Clostridium group; OC Bacillus/Staphylococcus group; Listeria. RN [1] RX MEDLINE; RA Haas A., Goebel W.; RT Cloning of a superoxide dismutase gene ; RL Mol. Gen. Genet. 231: (1992). FH Key Location/Qualifiers. FH. FT source FT /db_xref= taxon:1638 FT /organism= Listeria ivanovii FT /strain= ATCC FT RBS FT /gene= sod FT terminator FT CDS FT /db_xref= SWISS-PROT:P28763 FT /transl_table=11. EMBL-Record:")
48
ASN.1-Record: (Abstract Syntax Notation)
seq-set { seq { id { local str "VCREGA" } , descr { title "Volvox carteri f. nagariensis regA gene, genomic locus" , molinfo { biomol genomic } , create-date std { year 1998 , month 11 , day 16 } } , inst { repr raw , mol dna , length , seq-data ncbi2na 'FB07EFB13EDBE6FA215F5C3E07BF010CE891D3257E7306CD7E7BD F2F116F887486DE2BFBA54841CFF264F52F3F7823C07F2F8CA4E6FA9E7A7C5D9DB B 41B69C81FE8094CF2FF52801D411F243A6CD7E717E03F9E7A07A041BA2CF992F40ACAB416919AD .

49
Annotation Aufgaben: Umwandlung in Datenbankformate
Veröffentlichung der Sequenzdaten Kommentierung Verbindung mit weiteren Informationen z. B. Genstrukturen, regulatorische Elemente

50
Annotation Automatische Annotation im Rahmen von Sequenzprojekten
Manuelle Annotation: Überprüfung der automatisch generierten Daten Verbindung von: Gen-Vorhersage (codierender Bereich) Promotor- und enhancer-Vorhersage Datenbankvergleiche (homologe Sequenzen), EST-Datenbanken
 Promotor- und enhancer-Vorhersage. Datenbankvergleiche (homologe Sequenzen), EST-Datenbanken.")
51
Genstruktur-Vorhersage
Man unterscheidet zwischen Consensus und Nonconsensus (ab initio) Programmen. Consensus Methoden werden mittels eines bestimmten Satzes an Genen „trainiert“, codierende Bereiche zu finden. Diese Consensus Methoden sind sehr erfolgreich, wenn es sich um Gene handelt, die den Genen, mit welchen diese Programme trainiert worden sind, ähneln. Ab initio-Algorithmen versuchen hingegen, anhand grundlegender Charakteristika Gene zu finden
 Programmen. Consensus Methoden werden mittels eines bestimmten Satzes an Genen „trainiert , codierende Bereiche zu finden. Diese Consensus Methoden sind sehr erfolgreich, wenn es sich um Gene handelt, die den Genen, mit welchen diese Programme trainiert worden sind, ähneln. Ab initio-Algorithmen versuchen hingegen, anhand grundlegender Charakteristika Gene zu finden.")
52
Genstruktur-Vorhersage
Ansatzpunkte für ab initio – Algorithmen: Exon-Intron-Strukturen (Spleiß-Stellen, Pyrimidin-reiche Regionen am 3'-Ende von Introns etc.; GT-AG-Regel) statistische Auffälligkeiten in kodierenden Regionen GC-Gehalt, Codon usage ...
 statistische Auffälligkeiten in kodierenden Regionen. GC-Gehalt, Codon usage. ...")
53
Genstruktur-Vorhersage
Auffälligkeiten in kodierenden Regionen: 1. Positionsabhängige Nukleotidzusammensetzung: Beispiel: Das Testcode-Programm im gcg-Paket: Fickett‘s Statistik (1982) A-Position = Max(n(1), n(2), n(3)) / Min(n(1), n(2), n(3)) wobei n(1), n(2) und n(3) die Häufigkeit von A an den Positionen (1,4,7,...), (2,5,8,...) und (3,6,9,...) darstellen. Berechnung dieses Werts für alle 4 Basen in einem Sequenzfenster (>200 bp): A-position, C-position etc. und Verrechnung mit weiteren statistischen Parametern nichtcod. Seq: zufällige Verteilung der Basen: Position-Wert ~ 1 codierende Seq: gehäuftes Auftreten an best. Positionen: Position-Wert > 1
 A-Position = Max(n(1), n(2), n(3)) / Min(n(1), n(2), n(3)) wobei n(1), n(2) und n(3) die Häufigkeit von A an den Positionen (1,4,7,...), (2,5,8,...) und (3,6,9,...) darstellen. Berechnung dieses Werts für alle 4 Basen in einem Sequenzfenster (>200 bp): A-position, C-position etc. und Verrechnung mit weiteren statistischen Parametern. nichtcod. Seq: zufällige Verteilung der Basen: Position-Wert ~ 1. codierende Seq: gehäuftes Auftreten an best. Positionen: Position-Wert > 1.")
54
Testcode Exon Exon

55
Genstruktur-Vorhersage
2. Potentielle offene Leserahmen: Frames Exon Exon

56
Genstruktur-Vorhersage
3. Höherer GC-Gehalt an der dritten Stelle eines Codons: GC-Bias Exon Exon

57
Genstruktur-Vorhersage
4. Organismus-abhängige Codon-Auswahl: Codon usage / Codon Preference Codon usage table / Codon frequency table CUTG ID: Volvox_carteri_pl SPECIES: Volvox carteri SECTION: Plants SEQUENCES: 35 CODONS: AmAcid Codon Number / Fraction .. Gly GGG Gly GGA Gly GGT Gly GGC Glu GAG Glu GAA Asp GAT Asp GAC

58
Genstruktur-Vorhersage
4. Organismus-abhängige Codon-Auswahl: Codon usage / Codon Preference Gribskov et al, 1984 Bewertung der Häufigkeit einzelner Codons aus der analysierten Sequenz im Vergleich zur Codon Usage des Organismus Berechnung in einem Fenster (25 codons), das mit einem Inkrement von 3 Basen über die Sequenz geschoben wird Zusätzlich werden seltene Codons markiert
, das mit einem Inkrement von 3 Basen über die Sequenz geschoben wird. Zusätzlich werden seltene Codons markiert.")
59
Genstruktur-Vorhersage
4. Organismus-abhängige Codon-Auswahl: Codon usage / Codon Preference Exon Exon

60
Genstruktur-Vorhersage
Testansatz von Fickett & Tung, 1992: - GenBank unterteilt in 108 bp-Fenster ohne Überlappungen - nur die Fenster weiterverwenden, die entweder komplett kodierend oder komplett nicht-kodierend sind - Analyse der positionsabhängigen Nukleotid-Zusammensetzung, GC-Bias, Codon usage mit verschiedenen Algorithmen (mehr als 20 damals publizierte Methoden) -> Schlußfolgerung: einfachste Algorithmen, z. B. Oligomer-Zusammensetzung, sind effektiver (sensitiver und spezifischer) als viele andere Methoden
 -> Schlußfolgerung: einfachste Algorithmen, z. B. Oligomer-Zusammensetzung, sind effektiver (sensitiver und spezifischer) als viele andere Methoden.")
61
Genstruktur-Vorhersage
Ablauf von "Gene Finding"-Programmen: 1. Maskierung repetitiver DNA-Elemente: SINE, LINE, Organismus-spezifische wie z. B. Alu bei Primaten ... 2. Homologiesuche: BLAST gegen Datenbanken, u. a. EST - Protein-kodierende Gene über BLASTX - rRNA-Gene - tRNA-Gene - snRNA-Gene } über BLASTN

62
Genstruktur-Vorhersage
Ablauf von "Gene Finding"-Programmen: 3. Anwendung spezieller Suchparameter: - Codon usage - GC-Gehalt - offene Leseraster - Speißstellen - Nachbarschaft zu regulatorischen Elementen ... 4. Abgleich der gefundenen „Gene“ untereinander: - keine Überlappung von Genen - es gibt aber auch Ausnahmen, gerade bei Bakterien!

64
Definitionen: AP: tatsächlich positive Positionen, d. h. im Gen AN: tatsächlich negative Positionen, d. h. nicht im Gen PP: vorhergesagt (predicted) positive Positionen PN: vorhergesagt (predicted) negative Positionen TP: wahr positive Ausgaben TN: wahr negative Ausgaben FP: falsch positive Ausgaben FN: falsch negative Ausgaben Vorhersage-Genauigkeit: - Sensitivität: Sn = TP / AP (wieviele richtige überhaupt erkannt) - Spezifität: Sp = TP / PP (wieviele der richtig vorhergesagten sind wirklich richtig) - Selektivität: Sl = TN / AN (wieviele falsche werden als solche richtig erkannt) - Durchschnittliche Korrelation (approx. Correlation): AC = ((TP/(TP+FN)) + (TP/(TP+FP)) + (TN/(TN+FP)) + (TN/(TN+FN))) / 2 - 1
 positive Positionen. PN: vorhergesagt (predicted) negative Positionen. TP: wahr positive Ausgaben. TN: wahr negative Ausgaben. FP: falsch positive Ausgaben. FN: falsch negative Ausgaben. Vorhersage-Genauigkeit: - Sensitivität: Sn = TP / AP (wieviele richtige überhaupt erkannt) - Spezifität: Sp = TP / PP (wieviele der richtig vorhergesagten sind wirklich richtig) - Selektivität: Sl = TN / AN (wieviele falsche werden als solche richtig erkannt) - Durchschnittliche Korrelation (approx. Correlation): AC = ((TP/(TP+FN)) + (TP/(TP+FP)) + (TN/(TN+FP)) + (TN/(TN+FN))) /")
65
Genstruktur-Vorhersage

66
Genstruktur-Vorhersage

67
Genstruktur-Vorhersage

68
Genstruktur-Vorhersage
Ab initio - Vorhersagen: GENSCAN (1997, Christopher Burge, Prinzip: Fourier – Transformation. Auswertung der 3 – Basen Periodizität, welche codierende Bereiche aufweisen - ursprünglich entwickelt für menschliche und Vertrebraten-Sequenzen - strikt Organismus-spezifisch: Mais, Arabidopsis und Drosophila getestet; für Caenorhabditis in Entwicklung (Testphase) - ME: Missing Exons; WE: Wrong Exons
 - ME: Missing Exons; WE: Wrong Exons.")
69
Genstruktur-Vorhersage
Ab initio - Vorhersagen: GLIMMER (Gene Locator and Interpolated Markov Modeler, TIGR) Prinzip: Interpoliertes Markov Modell (IMM) für Bakterien - hohe Spezifität und Sensitivität verwendet u.a. bei der Genom-Sequenzierung von Borrelia burgdorferi, Thermotoga maritima oder Mycobacterium tuberculosis.
 Prinzip: Interpoliertes Markov Modell (IMM) für Bakterien - hohe Spezifität und Sensitivität. verwendet u.a. bei der Genom-Sequenzierung von Borrelia burgdorferi, Thermotoga maritima oder Mycobacterium tuberculosis.")
70
Genstruktur-Vorhersage
Neuere Ansätze nutzen zusätzlich EST-Daten: Spliced Alignment zur Exon-Intron-Vorhersage Beispiel: GeneSeqer jeder Treffer im Alignment erhält Exon-Status Introns sind lange Lücken im Alignment Splice site consensus wird berücksichtigt Scoring ergibt sich aus Alignment und Splice sites

71
Genstruktur-Vorhersage
GeneSeqer: Genomische DNA (z. B. BAC) als Query-Sequenz große Anzahl von ESTs Vorauswahl der ESTs durch "schmutzige" Alignments nur "gute" ESTs werden vollständig prozessiert spezielles EST-Format (dbEST) zum schnellen Zugriff Splice-Parameter für: human, mouse, rat, chicken, Drosophila, nematode, yeast, Aspergillus, Arabidopsis, maize Ursprünglich entwickelt für Arabidopsis
 als Query-Sequenz. große Anzahl von ESTs. Vorauswahl der ESTs durch schmutzige Alignments. nur gute ESTs werden vollständig prozessiert. spezielles EST-Format (dbEST) zum schnellen Zugriff. Splice-Parameter für: human, mouse, rat, chicken, Drosophila, nematode, yeast, Aspergillus, Arabidopsis, maize. Ursprünglich entwickelt für Arabidopsis.")
72
Genstruktur-Vorhersage
GeneSeqer: EST-Datenbanken Last update: March 12, 2001 Label Species # of ESTs soybean Glycine max , Drosophila Drosophila melanogaster , Arabidopsis Arabidopsis thaliana , C.elegans Caenorhabditis elegans , tomato Lycopersicon esculentum , M.truncatula Medicago truncatula , maize Zea mays , rice Oryza sativa , barley Hordeum vulgare , wheat Triticum aestivum , sorghum Sorghum bicolor , cotton Gossypium arboreum & hirsutum 36, pine Pinus taeda , L.japonicus Lotus japonicus , potato Solanum tuberosum , iceplant Mesembryanthemum crystallinum 14,033

73
Genstruktur-Vorhersage
SplicePredictor: trainiertes System zur Spleißstellenerkennung * Trainingssequenzen: Arabidopsis und Mais * Kennzeichen für Exon-Intron-Exon-Übergänge: - 5' GT - AG 3' - cAG an 3' - Pyrimidin-Stretch am 3'-Ende ( bp vor AG; >70 %) - Aufrechterhaltung des ORF
 - Aufrechterhaltung des ORF.")
74
Genstruktur-Vorhersage
SplicePredictor: trainiertes System zur Spleißstellenerkennung Example: t q loc sequence P rho gamma * P*R*G* parse ... A < gtatcagattggcAGtc (1 1 1) IIIAEEE-E-EDAEEEE D -> gagGTcttt (1 1 1) IIAEEEE-E-DAEEEEE D ----> gagGTaaca (3 4 4) IAEEEEE-D-AEEEEEE A < tttttcatatttcAGga (5 5 5) AEEEEED-A-EEEEEED A < atcagacgatttcAGgg (1 1 1) IIAEEDA-E-EEEEEDA
 IIIAEEE-E-EDAEEEE. D -> gagGTcttt (1 1 1) IIAEEEE-E-DAEEEEE. D ----> gagGTaaca (3 4 4) IAEEEEE-D-AEEEEEE. A < tttttcatatttcAGga (5 5 5) AEEEEED-A-EEEEEED. A < atcagacgatttcAGgg (1 1 1) IIAEEDA-E-EEEEEDA.")
75
Datenbanksuchen Vorhanden: unbekannte DNA- oder Proteinsequenz
Gesucht: gibt es diese oder eine ähnliche Sequenz in der Datenbank? Prinzip: Vergleich der Suchsequenz mit jeder einzelnen Sequenz in der Datenbank Bewertung der Ähnlichkeit anhand eines scoring-Algorithmus Ausgabe der Treffer mit dem besten score Problem: Optimale Algorithmen sind zu zeitaufwendig Heuristische Ansätze sind erforderlich

76
Datenbanksuchen: FASTA
Ablauf: Sequenzen der Datenbank werden mehrmals mit der Suchsequenz verglichen, zunächst grob, dann mit feineren Methoden. Lokale Alignments werden erstellt, um homologe Regionen zu finden. In jedem Durchlauf werden nur die möglicherweise Homologen behalten. Im Detail: FASTA ist ein Zwei-Schritt-Algorithmus mit vier Phasen: 1. Wortsuche zum Finden ähnlicher Regionen / Bewertung / Verbindung der Teile 2. Smith-Waterman-Alignment an diesen Regionen

77
Datenbanksuchen: FASTA
Für die Wortsuche werden Suchsequenz und Datenbank indiziert. Bei Proteinen wird eine Wortlänge von 2 und bei DNA von 6 verwendet Word List für FASTA, Word Size = 6 g c t g g a a g g c a t g c t g g a c t g g a a t g g a a g g g a a g g g a a g g c a a g g c a a g g c a t

78
Einschub: Hash-Verfahren
Die naive Suche eines Datensatzes in einer Liste dauert sehr lange Schnelle Suche mittels einer Hash-Funktion: h: K A K: Menge aller Schlüssel (Suchworte) A: Menge der Speicheradressen D.h. aus dem Suchwort (Schlüssel) wird direkt die Speicheradresse des Datensatzes berechnet.
 A: Menge der Speicheradressen. D.h. aus dem Suchwort (Schlüssel) wird direkt die Speicheradresse des Datensatzes berechnet.")
79
Datenbanksuchen: FASTA
Mit diesen k-tupeln mit der Länge 2 bzw. 6 werden exakte Treffer ermittelt. Diagonalfolgen liegen auf einer gedachten Matrix auf einer Diagonalen. Matches und Mismatches, aber keine Gaps!

80
Datenbanksuchen: FASTA
Innerhalb der 10 Diagonalfolgen mit den höchsten Scores werden lokale optimale Alignments bestimmt. Verwendet werden scoring-Matrizen (PAM oder BLOSUM) Der größte Score-Wert wird als init1 ausgegeben.
 Der größte Score-Wert wird als init1 ausgegeben.")
81
Datenbanksuchen: FASTA
Verlängerung der initialen Regionen zu größeren Alignments. Hier werden das erste Mal Lücken eingeführt, wenn nötig. Das erhaltene Alignment initn hat den maximalen Score unter Berücksichtigung der scoring-Matrix und der Gap Penalities.

82
Datenbanksuchen: FASTA
Ein zu initn alternativer Score opt wird errechnet. - Hierbei wird nur ein schmaler Streifen der Matrix ausgewertet. - Die Mitte ist durch init1 definiert. - Breite ist abhängig von der Wortlänge, z. B. 16 für ktup = 2 - Berechnung des opt-Wertes über Smith-Waterman - Verwendung von scoring-Matrix und Gap Penalties

83
Datenbanksuchen: BLAST
BLAST: Basic Local Alignment Search Tool BLAST ist ebenfalls eine Annäherung an den Smith-Waterman-Algorithmus. BLAST beginnt mit der Lokalisierung kurzer Teilsequenzen: Segment-Paare / hits Lokale optimale Paare, die je einen hit beinhalten, werden als HSPs (High-Scoring Segment-Pairs) bezeichnet. Beginn und Ende der HSPs wird so gewählt, daß eine Verkürzung oder Verlängerung den Score erniedrigen würde.
 bezeichnet. Beginn und Ende der HSPs wird so gewählt, daß eine Verkürzung oder Verlängerung. den Score erniedrigen würde.")
84
Datenbanksuchen: BLAST
Ablauf: Präprozessierung: Aus der Eingabesequenz wird die Menge aller Teilworte TW mit Länge w gebildet. Standard: Proteine: w=3, DNA: w=11 Teilwort B TW dient zur Bestimmung sämtlicher Worte (w-mere) mit Score S > T. Liste aller w-mers der Länge 2 mit Score S > T = 8 (BLOSUM 62) für die Sequenz RQCSAGW Teilwort B w-mers RQ RQ QC QC, RC, EC, NC, DC, HC, KC, MC, SC CS CS, CA, CN, CD, CQ, CE, CG, CK, CT SA kein w-mer der Länge 2 hat einen Score > 8 AG AG GW GW, AW, RW, NW, DW, QW, EW, HW, KW, PW, SW, TW, WW
 mit Score S > T. Liste aller w-mers der Länge 2 mit Score S > T = 8 (BLOSUM 62) für die Sequenz RQCSAGW. Teilwort B w-mers RQ RQ. QC QC, RC, EC, NC, DC, HC, KC, MC, SC. CS CS, CA, CN, CD, CQ, CE, CG, CK, CT. SA kein w-mer der Länge 2 hat einen Score > 8. AG AG. GW GW, AW, RW, NW, DW, QW, EW, HW, KW, PW, SW, TW, WW.")
85
Datenbanksuchen: BLAST
2. Lokalisierung der hits: Vergleichssequenz aus der Datenbank wird auf das Vorkommen der w-mere hin untersucht. Von jedem hit wird die Position bestimmt. Darstellung in einer Matrix (vgl. DotPlot). hit
. hit.")
86
Datenbanksuchen: BLAST
3. Bestimmung der HSPs: Welche Paare von hits liegen auf einer Diagonale der Matrix? Berücksichtigung des räumlichen Abstands A der hits. Für Proteine wird A = 40 gewählt. hit mit Abstand < A

87
Datenbanksuchen: BLAST
4. Erweiterung mit Lücken: In einer gedachten Matrix wird das Alignment in beide Richtungen verlängert. Parameter Xg begrenzt die Verlängerung durch minimalen Score, der angenommen werden darf. Lücken sind erlaubt. Abweichung des Scores < Xg

88
Datenbanksuchen: Vergleich FASTA - BLAST
BLAST ist: schneller sensitiver bei Proteinsuchen, da sequenzähnliche Oligomere verwendet werden statt identische Dipeptide bei FASTA flexibler, da Nukleotidsequenzen in alle 6 Leserahmen umgesetzt werden können FASTA ist: sensitiver bei Nukleotidsuchen, da kürzere (6 statt 11) Wortlängen verwendet werden besser geeignet im Vergleich cDNA gegen genomische Datenbanken -> bei Gap Extension Penalty = 0 können auch lange Introns übersprungen werden -> BLAST würde nur das längste Exon finden (wenn überhaupt)
 Wortlängen verwendet werden. besser geeignet im Vergleich cDNA gegen genomische Datenbanken -> bei Gap Extension Penalty = 0 können auch lange Introns übersprungen werden -> BLAST würde nur das längste Exon finden (wenn überhaupt)")
89
Datenbanksuchen: BLAST
Verschiedene BLAST-Programme: blastn: Nukleotidsequenz gegen Nukleotiddatenbank blastp: Proteinsequenz gegen Proteindatenbank blastx: translatierte Nukleotidsequenz (alle 6 Leserahmen) gegen Proteindatenbank tblastn: Proteinsequenz gegen translatierte Nukleotiddatenbank tblastx: translatierte Nukleotidsequenz (alle 6 Leserahmen) gegen translatierte Nukleotiddatenbank (alle 6 Leserahmen)
 gegen Proteindatenbank. tblastn: Proteinsequenz gegen translatierte Nukleotiddatenbank. tblastx: translatierte Nukleotidsequenz (alle 6 Leserahmen) gegen translatierte Nukleotiddatenbank (alle 6 Leserahmen)")
90
Datenbanksuchen: Statistische Signifikanz
Wahrscheinlichkeits-Dichtefunktion (Extremwertverteilung) Bezug eines erhaltenen Alignment-Scores S zur erwarteten Verteilung: - P-Wert: Maß für die Wahrscheinlichkeit, daß ein Alignment mit dem Score S oder besser durch reinen Zufall entstünde (gut: P gegen 0) - E-Wert: Erwartete Anzahl von zufälligen Alignments mit Scores S Signifikanz abhängig von der Größe des gesamten Suchraums (z. B. Anzahl der Aminosäuren/Nukleotide in der Datenbank) und der erwarteten Länge des lokalen Alignments Exakte statistische Theorie existiert nur für Alignments ohne Lücken.
 Bezug eines erhaltenen Alignment-Scores S zur erwarteten Verteilung: - P-Wert: Maß für die Wahrscheinlichkeit, daß ein Alignment mit dem Score S. oder besser durch reinen Zufall entstünde (gut: P gegen 0) - E-Wert: Erwartete Anzahl von zufälligen Alignments mit Scores S. Signifikanz abhängig von der Größe des gesamten Suchraums. (z. B. Anzahl der Aminosäuren/Nukleotide in der Datenbank) und der erwarteten Länge des lokalen Alignments. Exakte statistische Theorie existiert nur für Alignments ohne Lücken.")
91
Datenbanksuchen: Statistische Signifikanz
Frage: Ist ein bestimmtes Alignment mit einem Score S ein Beweis für die Homologie? Abschätzung des Erwartungswertes durch Zufallsalignments. 3 Möglichkeiten des Zufallmodells: 1. echte, aber nicht-homologe Sequenzen 2. echte Sequenzen, aber in ihrer Abfolge permutiert -> Zusammensetzung ist beibehalten Beispiel: Originalsequenz: ACGTACGT Permutierte Seq: ACGTACTG TGCATGCA usw. 3. zufällig erzeugte Sequenzen, evtl. unter Berücksichtigung eines Modells (z. B. Häufigkeitsverteilung der Aminosäuren)
")
92
Alignments: Statistische Signifikanz
Statistik zu lokalen Alignments ohne Lücken: - HSPs sind lokal optimal, weisen einen Score S auf und haben keine Lücken - Statistik möglich bei genügend langen Sequenzen (Längen n und m) (n: Länge der Suchsequenz, m: Länge der Datenbanksequenz) - Zwei Parameter für den Suchraum und das Scoring-System: K und l Erwartete Anzahl von HSPs mit Scores S: E = Kmne-lS E-Wert (E-value) für den beobachteten Score S eines HSPs -> Verdoppelung der Sequenzlänge verdoppelt die Anzahl von zufälligen HSPs mit Scores S
 (n: Länge der Suchsequenz, m: Länge der Datenbanksequenz) - Zwei Parameter für den Suchraum und das Scoring-System: K und l. Erwartete Anzahl von HSPs mit Scores S: E = Kmne-lS. E-Wert (E-value) für den beobachteten Score S eines HSPs. -> Verdoppelung der Sequenzlänge verdoppelt die Anzahl von zufälligen HSPs. mit Scores S.")
93
S‘ und E werden in der BLAST-Ausgabe angegeben
Alignments: Statistische Signifikanz Reine Score-Werte S geben keine Information über die Qualität des Ergebnisses ohne genaue Kenntnis über die Datenbank und den Suchalgorithmus (bzw. K und l) (vergleichbar mit Längenangabe ohne Einheit, z. B > Meter, Kilometer, Lichtjahre ...) -> Bit-Scores S' zur Normalisierung (auf Suchraum und Scoring-System): S' = (lS - lnK) / ln2 aus den Bit-Scores S' lassen sich E-Werte ableiten, die nur von den Sequenzlängen abhängen: E = mn2-S' -> für die Signifikanz-Beurteilung sind dann nur noch die Sequenzlängen m und n zu wissen S‘ und E werden in der BLAST-Ausgabe angegeben
 (vergleichbar mit Längenangabe ohne Einheit, z. B > Meter, Kilometer, Lichtjahre ...) -> Bit-Scores S zur Normalisierung (auf Suchraum und Scoring-System): S = (lS - lnK) / ln2. aus den Bit-Scores S lassen sich E-Werte ableiten, die nur von den Sequenzlängen abhängen: E = mn2-S -> für die Signifikanz-Beurteilung sind dann nur noch die Sequenzlängen m und n zu wissen. S‘ und E werden in der BLAST-Ausgabe angegeben.")
94
Alignments: Statistische Signifikanz
Die Wahrscheinlichkeit, exakt a HSPs mit Score S zu finden (Poisson-Verteilung): P = e-E(Ea/a!) Für mind. 1 HSP mit einem Score S gilt: P = 1 - e-E Der P-Wert ist auf diese Weise mit dem beobachteten Score S gekoppelt. Für E < 0,01 gilt: P E für größere Werte wird E jedoch deutlich größer, was für den Anwender anschaulicher ist P: E: 5 10
: P = e-E(Ea/a!) Für mind. 1 HSP mit einem Score S gilt: P = 1 - e-E. Der P-Wert ist auf diese Weise mit dem beobachteten Score S gekoppelt. Für E < 0,01 gilt: P E. für größere Werte wird E jedoch deutlich größer, was für den Anwender anschaulicher ist. P: E:")
95
Datenbanksuchen: Statistische Signifikanz
Speziell für Datenbanksuchen gilt: Signifikanz muß berechnet werden für einen Vergleich eines Proteins mit Länge m gegen eine Datenbank mit vielen Proteinen unterschiedlicher Länge 1. Möglichkeit: a priori-Annahme, daß alle Proteine gleich wahrscheinlich mit der Query-Sequenz verwandt sind. E-Wert ergibt sich aus dem Produkt des E-Werts eines Pairwise-Alignments mit der Anzahl der Proteine in der durchsuchten Datenbank. (FASTA für Proteine nutzt diese Berechnung)
")
96
Datenbanksuchen: Statistische Signifikanz
2. Möglichkeit: a priori-Annahme, daß Query-Sequenz mit höherer Wahrscheinlichkeit zu längeren Sequenzen verwandt ist. Begründet wird dies mit der Domänenstruktur der Proteine. Der E-Wert eines Pairwise-Alignments wird multipliziert mit dem Faktor N/n, wobei N: "Länge" der Datenbank in Nukleotiden bzw. Aminosäureresten und n: Länge der verglichenen Datenbanksequenz. Wird z. B. von BLAST verwendet (E-Wert bei Ausgabe der Suchergebnisse). aus E = Kmne-lS wird dann E = KmNe-lS Bzw. aus E = mn2-S wird dann E = mN2-S'
. aus E = Kmne-lS wird dann E = KmNe-lS. Bzw. aus E = mn2-S wird dann E = mN2-S")
97
Datenbanksuchen: Statistische Signifikanz
Statistiken für Alignments mit Lücken: -> Abschätzen der Parameter aus vielen Vergleichen FASTA: echte Sequenzen, kein Zufallsmodell -> optimale Scores (lokal!) für Query-Sequenz gegen jede Datenbanksequenz -> l und K bestimmbar BLAST: Vorabschätzung von l und K durch Zufallsmodell -> schneller, da optimale lokale Scores nur aus Vergleich mit ein paar unverwandten Sequenzen -> zusätzlich in BLAST Korrektur der "Kanten-Effekte"
 für Query-Sequenz gegen jede Datenbanksequenz. -> l und K bestimmbar. BLAST: Vorabschätzung von l und K durch Zufallsmodell. -> schneller, da optimale lokale Scores nur aus Vergleich. mit ein paar unverwandten Sequenzen. -> zusätzlich in BLAST Korrektur der Kanten-Effekte")
98
Datenbanksuchen: Praktischer Ablauf am Beispiel BLAST
Adresse: Verwendbare Formate: 1. FASTA-Format: Beinhaltet in der 1. Zeile nach dem ">"-Zeichen eine Sequenzbeschreibung, anschließend die Sequenz ohne Unterbrechungen. Das Ende der Sequenz wird nach zwei Leerzeichen automatisch erkannt. Beispiel: >gi|129295|sp|P01013|OVAX_CHICK GENE X PROTEIN (OVALBUMIN-RELATED) QIKDLLVSSSTDLDTTLVLVNAIYFKGMWKTAFNAEDTREMPFHVTKQESKPVQMMCMNNSFNVATLPAE KMKILELPFASGDLSMLVLLPDEVSDLERIEKTINFEKLTEWTNPNTMEKRRVKVYLPQMKIEEKYNLTS VLMALGMTDLFIPSANLTGISSAESLKISQAVHGAFMELSEDGIEMAGSTGVIEDIKHSPESEQFRADHP FLFLIKHNPTNTIVYFGRYWSP
 QIKDLLVSSSTDLDTTLVLVNAIYFKGMWKTAFNAEDTREMPFHVTKQESKPVQMMCMNNSFNVATLPAE. KMKILELPFASGDLSMLVLLPDEVSDLERIEKTINFEKLTEWTNPNTMEKRRVKVYLPQMKIEEKYNLTS. VLMALGMTDLFIPSANLTGISSAESLKISQAVHGAFMELSEDGIEMAGSTGVIEDIKHSPESEQFRADHP. FLFLIKHNPTNTIVYFGRYWSP.")
99
Datenbanksuchen: Praktischer Ablauf am Beispiel BLAST
2. Reine Sequenzen: Wie FASTA-Format, aber ohne Sequenzbeschreibung. Beispiel: QIKDLLVSSSTDLDTTLVLVNAIYFKGMWKTAFNAEDTREMPFHVTKQESKPVQMMCMNNSFNVATLPAE KMKILELPFASGDLSMLVLLPDEVSDLERIEKTINFEKLTEWTNPNTMEKRRVKVYLPQMKIEEKYNLTS VLMALGMTDLFIPSANLTGISSAESLKISQAVHGAFMELSEDGIEMAGSTGVIEDIKHSPESEQFRADHP FLFLIKHNPTNTIVYFGRYWSP

100
Datenbanksuchen: Praktischer Ablauf am Beispiel BLAST
3. GenBank-Flatfile-Sequenz / gcg-Format ohne Header: Üblicherweise aus GenBank-Ausgabe bzw. aus dem gcg-Format kopiert, wobei keine Informationen außer Zählungshilfen vorhanden sein dürfen. Leerzeilen sind zu entfernen, da sie für die Erkennung des Sequenzendes herangezogen werden. Beispiel: 1 QIKDLLVSSS TDLDTTLVLV NAIYFKGMWK TAFNAEDTRE MPFHVTKQES KPVQMMCMNN 61 SFNVATLPAE KMKILELPFA SGDLSMLVLL PDEVSDLERI EKTINFEKLT EWTNPNTMEK 121 RRVKVYLPQM KIEEKYNLTS VLMALGMTDL FIPSANLTGI SSAESLKISQ AVHGAFMELS 181 EDGIEMAGST GVIEDIKHSP ESEQFRADHP FLFLIKHNPT NTIVYFGRYW SP

101
Datenbanksuchen: Praktischer Ablauf am Beispiel BLAST
Beispielsequenz: Ein Prion? 1 MSLLAYWLAS LWVTMWTDVG LCKKRPKPGG WNTGGRRYPA DGSPGGNRYP PQGATWGQPY 61 GGGWGQPHGG SFGQPHGGSW GQPHAAAWGQ GGGTHNQWNK PSKPKTNLKH VAGAAAAGAV 121 VGGLGGYMLG SAMSRPMIHF GNDWEDRYYR ENMYRYPNQV YYRPVDQYSN QNNFVHDCVN 181 ITIKQHTVTT TTKGENFTET

102
Datenbanksuchen: Praktischer Ablauf am Beispiel BLAST

103
Datenbanksuchen: Praktischer Ablauf am Beispiel BLAST

104
Datenbanksuchen: Praktischer Ablauf am Beispiel BLAST

105
Datenbanksuchen: Praktischer Ablauf am Beispiel BLAST

106
Datenbanksuchen: Praktischer Ablauf am Beispiel BLAST

107
Datenbanksuchen: Praktischer Ablauf am Beispiel BLAST

108
Datenbanksuchen: Praktischer Ablauf am Beispiel BLAST

109
Datenbanksuchen: Praktischer Ablauf am Beispiel BLAST

110
Datenbanksuchen: Praktischer Ablauf am Beispiel BLAST

111
Datenbanksuchen: Praktischer Ablauf am Beispiel BLAST

112
Datenbanksuchen: Praktischer Ablauf am Beispiel BLAST

113
Datenbanksuchen: Praktischer Ablauf am Beispiel BLAST

Ähnliche Präsentationen







 auf.>")


>")




 = 5n 3 + n + 1000 für alle n a)Geben sie eine obere Schranke O(g(n)) an. b)Beweisen.>")





 Matrixkettenprodukt>")
 Prof. Th. Ottmann.>")
