Präsentation herunterladen
Die Präsentation wird geladen. Bitte warten

1
Klassifizierung elektroanalytischer Methoden
ohne Elektrodenreaktion mit Elektrodenreaktion FARADAYscher Strom = 0 FARADAYscher Strom 0 FARADAYscher Strom 0 Kennzeichen variables konstantes Potential Potential Konduktometrie Methode Potentiometrie Voltametrie/ Polarographie Amperometrie Meßgröße Leitfähigkeit Potentialdifferenz Stromstärke = f(U) Stromstärke; Masse
 Stromstärke; Masse.")
2
Elektrochemische Analysenmethoden
Leitfähigkeitsmessung (konduktometrisch) pH-Wert-Bestimmung (potentiometrisch) Ionenselektive Elektroden (potentiometrisch) Redoxpotentialmessung (potentiometrisch) Erfassung des gelösten Sauerstoffs (amperometrisch) Voltametrie/Polarographie Leitfähigkeit - summarische Methode - konduktometrische Bestimmung der Ionenstärke/gelösten Salze - keine Aussagen über Ionenarten Prinzip: - Zusammenhang Leitfähigkeit - Konzentration Elektrolyte - Messung Widerstand: WHEATSTON‘sche Brückenschaltung
 pH-Wert-Bestimmung (potentiometrisch) Ionenselektive Elektroden (potentiometrisch) Redoxpotentialmessung (potentiometrisch) Erfassung des gelösten Sauerstoffs (amperometrisch) Voltametrie/Polarographie. Leitfähigkeit. - summarische Methode. - konduktometrische Bestimmung der Ionenstärke/gelösten Salze. - keine Aussagen über Ionenarten. Prinzip: - Zusammenhang Leitfähigkeit - Konzentration Elektrolyte. - Messung Widerstand: WHEATSTON‘sche Brückenschaltung.")
3
L r R= A A = Elektrodenoberfläche L = Elektrodenabstand - Widerstand:
- elektrische Leitfähigkeit: - Problem: Polarisierung der Elektroden, scheinbare Erhöhung Widerstand Verwendung des Wechselstroms hoher Frequenz Verwendung der Größe Elektrodenoberfläche (Platinierung) Vier-Elektrodenmessung mit getrennten stromführenden und Spannungsmeßelektroden L R= A r A = Elektrodenoberfläche L = Elektrodenabstand ρ = spezifische Widerstand [mS/cm; µS/cm]
 Vier-Elektrodenmessung mit getrennten stromführenden und Spannungsmeßelektroden. L. R= A. r. A = Elektrodenoberfläche. L = Elektrodenabstand. ρ = spezifische Widerstand. [mS/cm; µS/cm]")
4
Einflussfaktoren auf die Größe der Leitfähigkeit:
Zahl der Ionen Ladung pro Ion Ionenbeweglichkeit Temperatur Beweglichkeit eines Ions hängt ab von: Ladung Ionenradius Verhältnis Ladung – Ionenradius Solvationshülle Lösungsmittel (Viskosität) + - Fe FR Katode Anode η
 + - Fe. FR. Katode. Anode. η.")
5
λ = λ∞ - a√c für starke Elektrolyte
Leitfähigkeit besonders hohen Beitrag zur Leitfähigkeit: H+ und OH- linearer Zusammenhang Äquivaentleitfähigkeit – Elektrolytkonzentration nur in sehr verdünnten Lösungen – KOHLRAUSCHschen Gesetz für Konzentration kleiner 10-2 mol/L ( λ = κ/c*ze→ F(u+ + u-) = λ+ + λ- λ = λ∞ - a√c für starke Elektrolyte λ = λ∞ – a`c für schwache Elektrolyte (Dissoziationsgrad) Ursachen: Wechselwirkungen, Dissoziationsgrad Ionenäquivalentleifähigkeit λ∞ in Scm2/mol in Abhängigkeit von der Temperatur Temperatur in °C 18 25 50 100 H+ 240 314 350 465 644 OH- 105 172 192 284 439 Na+ 26,0 43,5 50,9 82,0 155 Cl- 41,1 65,5 75,5 116 207
 = λ+ + λ- λ = λ∞ - a√c für starke Elektrolyte. λ = λ∞ – a`c für schwache Elektrolyte (Dissoziationsgrad) Ursachen: Wechselwirkungen, Dissoziationsgrad. Ionenäquivalentleifähigkeit λ∞ in Scm2/mol in Abhängigkeit von der Temperatur. Temperatur in °C H OH Na+ 26,0. 43,5. 50,9. 82, Cl- 41,1. 65,5. 75,")
6
Praxis Messtechnische Hinweise
(Leitfähigkeitsmessungen sind Widerstandsmessungen mit Wechselstrom) Kontrolle - elektrischer Kontakte auf Sauberkeit - Luftblasen im Raum zwischen der Elektroden Geräte besitzen in der Regel verschiedene Messbereiche (mS/cm; µS/cm) mit Messfrequenzanpassung und Temperaturkompensation Funktionsprüfung/Ermittlung Zellkonstante mit definierter KCl-Lösung Behandlung von Messzellen (gut Spülen, Pt/Pt- Aufbewahren im Wasser) Messung kleiner Leitfähigkeiten unter 10 µS/cm unter Luftabschluss
 Kontrolle - elektrischer Kontakte auf Sauberkeit. - Luftblasen im Raum zwischen der Elektroden. Geräte besitzen in der Regel verschiedene Messbereiche (mS/cm; µS/cm) mit Messfrequenzanpassung und Temperaturkompensation. Funktionsprüfung/Ermittlung Zellkonstante mit definierter KCl-Lösung. Behandlung von Messzellen (gut Spülen, Pt/Pt- Aufbewahren im Wasser) Messung kleiner Leitfähigkeiten unter 10 µS/cm unter Luftabschluss.")
7
Praxis Anwendung der Leitfähigkeitsmessung
hohe Leitfähigkeit ist Hinweis auf Verschmutzung, geologischer Einfluss oder Meerwassereintrag Korrelation Leitfähigkeit – Ionenbilanz Reinheitskontrolle von Wasser (Ionenaustausch: 0,01 µS/cm → 5 µg/L) Betriebskontrolle (Spülprozesse in Molkerei und bei der Bierherstellung) Konduktometrische Titration (H+ + Cl- + Na+ + OH- → H2O + Na+ + Cl-) µs/cm ms/cm 0,1 0, Speisewasser für Wasserrohrkessel Vollentsalzung Ionenaustauscher Regenwasser Trinkwasser Oberflächenwässer Meerwasser Industrielles Prozesswässer
 Betriebskontrolle (Spülprozesse in Molkerei und bei der Bierherstellung) Konduktometrische Titration (H+ + Cl- + Na+ + OH- → H2O + Na+ + Cl-) µs/cm. ms/cm. 0,1 0, Speisewasser für Wasserrohrkessel. Vollentsalzung Ionenaustauscher. Regenwasser. Trinkwasser. Oberflächenwässer. Meerwasser. Industrielles Prozesswässer.")
8
Grundlagen der Potentiometrie
Zn + - Zn2+ ZnCl2 ? - Entstehung einer Galvani-Spannung E0(1/2H2/H+) Elektrodenarten (je nach der Anzahl der Gleichgewichte unterscheidet man Elektroden 1., 2. und 3) Elektrode 1. Art ( ) Elektroden 2. Art (Ag/AgCl ) Elektroden 3. Art (Ag/Ag2S +PbS) Potentialmessung Messung der Spannung zwischen den beiden Elektroden immer stromlos Kein elektrischer Strom im Stromkreis
 Elektrodenarten (je nach der Anzahl der Gleichgewichte unterscheidet man Elektroden 1., 2. und 3) Elektrode 1. Art ( ) Elektroden 2. Art (Ag/AgCl ) Elektroden 3. Art (Ag/Ag2S +PbS) Potentialmessung. Messung der Spannung zwischen den beiden Elektroden immer stromlos. Kein elektrischer Strom im Stromkreis.")
9
Referenzelektroden (1)
Standardwasserstoffelektrode Ag/AgCl, Cl--Elektrode Hg/Hg2Cl2,Cl--Elektrode (Kalomelelektrode) Tl(Hg)/TlCl, Cl--Elektrode (Thalamidelektrode®) Hg/Hg2SO4, SO42--Elektrode Anforderungen an eine Referenzelektrode stabiles, reproduzierbares Potential (besser als 0.1 mV) für eine große Variationsbreite der Einsatzbedingungen (Redoxverhalten, pH, Temperatur) keine Wechselwirkung mit der Messelektrode keine Veränderung des Analyten durch Elektrolytlösung der Referenzelektrode.
 Tl(Hg)/TlCl, Cl--Elektrode (Thalamidelektrode®) Hg/Hg2SO4, SO42--Elektrode. Anforderungen an eine Referenzelektrode. stabiles, reproduzierbares Potential (besser als 0.1 mV) für eine große. Variationsbreite der Einsatzbedingungen (Redoxverhalten, pH, Temperatur) keine Wechselwirkung mit der Messelektrode. keine Veränderung des Analyten durch Elektrolytlösung der Referenzelektrode.")
10
Referenzelektroden (2)
Diaphragmaform von Bezugselektroden: Schliffdiaphragma, (b) Keramikdiaphragma, (c) Lochdiaphragma einer Einstabmesskette mit Polymermatrix-Füllung
 Keramikdiaphragma, (c) Lochdiaphragma. einer Einstabmesskette mit Polymermatrix-Füllung.")
11
Potentiometrische pH-Wert-Bestimmung
Bedeutung: allgemeiner Parameter Feststellung Gewässergüte (TrinkwV: pH= 6,5…9,5) Kriterium für Möglichkeit stattfindender Prozesse (z:B. 5FeS2 + 14NO3- + 8H2O = 7N2 + 10SO FeOOH + 11H+ 2FeS2 + 7O2 + 2H2O = 2Fe2+ + 4SO H+) Bedeutung für Optimierung vieler Verfahren der Wasseraufbereitung und Abwasserbehandlung Aussagen zu einzelnen Wasserinhaltsstoffen/Prozessen Prinzip: Messung: Potentialdifferenz Bezugselektrode – Messelektrode Bezugselektrode: konstantes, bekanntes Potential Messelektrode: Potential wird durch pH-Wert bestimmt Potentialbestimmender Prozess: Diffusions- und Ionenaustauschvorgang
 Kriterium für Möglichkeit stattfindender Prozesse. (z:B. 5FeS2 + 14NO3- + 8H2O = 7N2 + 10SO FeOOH + 11H+ 2FeS2 + 7O2 + 2H2O = 2Fe2+ + 4SO H+) Bedeutung für Optimierung vieler Verfahren der Wasseraufbereitung und Abwasserbehandlung. Aussagen zu einzelnen Wasserinhaltsstoffen/Prozessen. Prinzip: Messung: Potentialdifferenz Bezugselektrode – Messelektrode. Bezugselektrode: konstantes, bekanntes Potential. Messelektrode: Potential wird durch pH-Wert bestimmt. Potentialbestimmender Prozess: Diffusions- und Ionenaustauschvorgang.")
12
Sulfat- und Nitratkonzentrationen im oberflächennahen Grundwasser und Pyritgehalt im Sediment, Mockritz N River Elbe Brunnen Fließrichtung 1 km 200 300 500 700 1100 Wasser Sulfat in mg/L Elbe Wasserwerk Elbe Wasserwerk 2000 4000 6000 8000 N 150 mg/L NO3 200 mg/L NO3 1 km 0 mg/L NO3 Sediment Pyrit in mg/kg 5FeS2 + 14NO3- + 8H2O = 7N2 + 10SO FeOOH + 11H+

13
pH-Wert ΔU Einstabmesskette, Glas- und Bezugselektrode sind in einem System vereinigt. Lösung der Bezugselektrode (meist KCl) Gewässerprobe Bezugselektrode Diaphragma Pufferlösung mit konstanter [H3O]+ Glasmembran 72% SiO2 22% Na2O 6% CaO Anschlusselektrode

14
Potentiometrische pH-Wert-Bestimmung
Messelektrode (Glaselektrode): geschlossene Glaskugel mit Pufferlösung (definierter pH) spezielles dünnwandiges Glas, quellfähig, leitend (Gitterionen) Ausbildung „äußere“ und „innere“ Gelschicht (dort Ionenaustausch- und Diffusionsprozesse) in Probe: Bildung inneres und äußeres Potential Messung Potentialdifferenz (durch Ableiten innere und äußere Elektrode) NERNST: 25 °C:
: geschlossene Glaskugel mit Pufferlösung (definierter pH) spezielles dünnwandiges Glas, quellfähig, leitend (Gitterionen) Ausbildung „äußere und „innere Gelschicht (dort Ionenaustausch- und Diffusionsprozesse) in Probe: Bildung inneres und äußeres Potential. Messung Potentialdifferenz (durch Ableiten innere und äußere Elektrode) NERNST: 25 °C:")
16
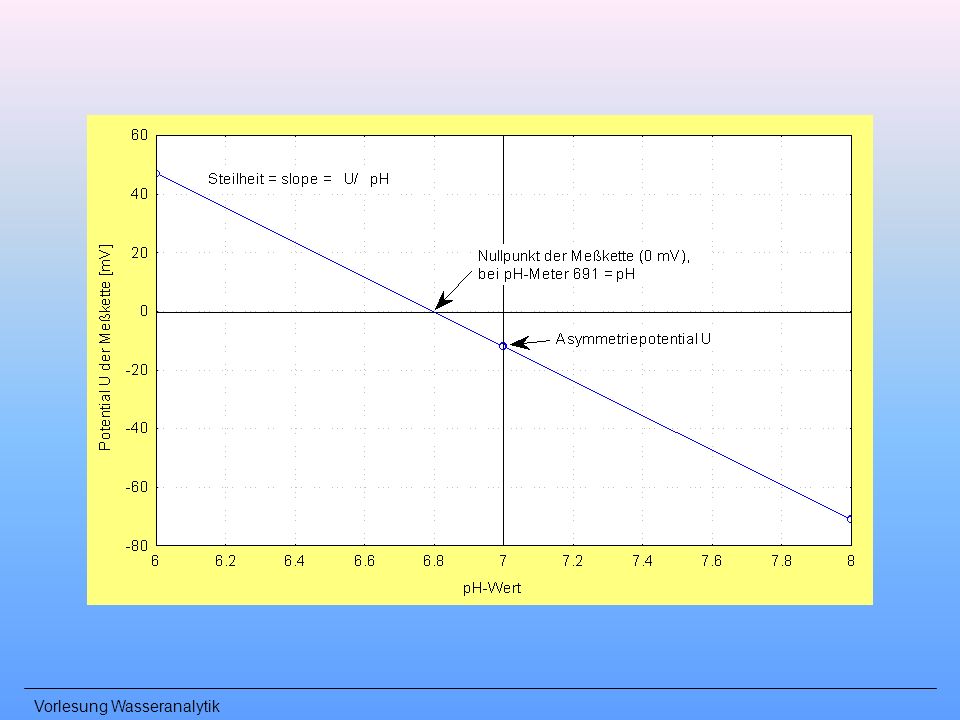
Potentiometrische pH-Wert-Bestimmung
Praxis: pH-Messung temperaturabhängig (NERNST), i. a. Temperatur kompensiert vor der ersten Benutzung: Quellen der Elektrode Einstabmessketten in KCl-Lösung aufbewahren Glaselektrode nicht trocken abreiben, nie erhitzen Messung in stark alkalischen Medien möglichst kurz öl-, fetthaltige Wässer: spezielle Reinigung (u. a. Ethanol) mindestens je Messtag kalibrieren (Steilheit)
, i. a. Temperatur kompensiert. vor der ersten Benutzung: Quellen der Elektrode. Einstabmessketten in KCl-Lösung aufbewahren. Glaselektrode nicht trocken abreiben, nie erhitzen. Messung in stark alkalischen Medien möglichst kurz. öl-, fetthaltige Wässer: spezielle Reinigung (u. a. Ethanol) mindestens je Messtag kalibrieren (Steilheit)")
17
pH-Wert Potentiometrie weiter ionenselektive Elektroden: (Festkörper- und Flüssigmembranelektroden) potentiometrische Verfahren Messelektrode: Innenraum bekannte Lösung, außen: Probelösung und Bezugselektrode dazwischen heterogene Membran oder kristalliner Festkörper (schwerlösliches, anorganisches Salz) an der Phasengrenze Messlösung – Membran (Festkörper) bilden Stoffe selektiv Potential aus (Funktion der Konzentration) Ziel: nur einzelne Ionenart messbar, in der Praxis oft Querempfindlichkeit Beispiele: fluorselektive Elektrode (Lanthanfluoridkristall) Festkörperelektroden für Silber-, Chlorid-, Bromid-, Iodid- und Sulfidmessungen
 an der Phasengrenze Messlösung – Membran (Festkörper) bilden Stoffe selektiv Potential aus (Funktion der Konzentration) Ziel: nur einzelne Ionenart messbar, in der Praxis oft Querempfindlichkeit. Beispiele: fluorselektive Elektrode (Lanthanfluoridkristall) Festkörperelektroden für Silber-, Chlorid-, Bromid-, Iodid- und Sulfidmessungen.")
18
pH-Wert Gegenüberstellung der 3 wichtigsten Bauformen ionenselektiver Elektroden 5 4 4 4 3 3 3 2 2 2 1 1 1 pH-Glaselektrode: 1 = Glasmembran 2 = innere Ableitung 3 = Innenpuffer 4 = Schaft 5 = Elektrodenkabel Ionenselektive Elektrode mit Festkörpermembran 1 = Membran 2 = innere Ableitung 3 = Innenlösung 4 = Schaft Ionenselektive Elektrode mit flüssiger Membran 1 = poröse Membran mit mess-technisch aktiver organischer Phase (3) getränkt 2 = innere Ableitung (wässrige Lösung) 3 = organische Phase (Vorrat) 4 = Schaft LaF3= LaF2+ + F- E = Econst - 59,16 lg aF E = Econst - 59,16 pH I2Ca = 2I- + Ca2+ E= Econst + 59,16/2 lg aCa pH=6…8
 getränkt. 2 = innere Ableitung (wässrige Lösung) 3 = organische Phase (Vorrat) 4 = Schaft. LaF3= LaF2+ + F- E = Econst - 59,16 lg aF. E = Econst - 59,16 pH. I2Ca = 2I- + Ca2+ E= Econst + 59,16/2 lg aCa. pH=6…8.")
19
Strukturformeln von Ionophorverbindungen

20
Übersicht über ionenselktiven Elektroden

21
Redoxpotential (Redoxspannung)
Redoxpotential bestimmt chemische und biologische Prozesse in Gewässern Grundlagen: Ox + ne- Red NERNSTsche Gleichung:

22
Redoxpotential (Redoxspannung)
Potentialerniedrigung bei Erhöhung Konzentration reduzierter Spezies Potentialerhöhung, je mehr oxidierte Stoffe vorhanden sind bei a (Ox) = a (Red) ist Eredox = E0 (Standardpotential) in Wasserproben i. a. Vielzahl Redoxsysteme, Messung der Summe der Redoxpotentiale Redoxsystem häufig unter Beteiligung von Protonen pH-Abhängigkeit temperaturabhängig (Temperatur immer angeben) neben Einzelpotential der Messelektrode (Platin) auch Berücksichtigung Bezugselektrode (z. B. Kalomelektrode)
 = a (Red) ist Eredox = E0 (Standardpotential) in Wasserproben i. a. Vielzahl Redoxsysteme, Messung der Summe der Redoxpotentiale. Redoxsystem häufig unter Beteiligung von Protonen pH-Abhängigkeit. temperaturabhängig (Temperatur immer angeben) neben Einzelpotential der Messelektrode (Platin) auch Berücksichtigung Bezugselektrode (z. B. Kalomelektrode)")
23
Redoxpotential (Redoxspannung)
Praxis: regelmäßig Messkette überprüfen (gesättigte Chinhydronlösung in Pufferlösung) Größe des Redoxpotentials abhängig von: pH-Wert Temperatur Ionenaktivität Beschaffenheit Elektrodenoberfläche Störungen durch Beläge auf Platinelektrode (z. B. Manganoxidhydrat) lange Einstellzeiten beim Wechsel von Lösungen mit verschiedenen Ionenaktivitäten Redoxpotential: Anhaltspunkt für aerobe/anaerobe Prozesse: < -200 mV: strikt anaerob -200 bis 0 mV: Übergangscharakter positive Werte: aerobe Vorgänge
 Größe des Redoxpotentials abhängig von: pH-Wert. Temperatur. Ionenaktivität. Beschaffenheit Elektrodenoberfläche. Störungen durch Beläge auf Platinelektrode (z. B. Manganoxidhydrat) lange Einstellzeiten beim Wechsel von Lösungen mit verschiedenen Ionenaktivitäten. Redoxpotential: Anhaltspunkt für aerobe/anaerobe Prozesse: < -200 mV: strikt anaerob bis 0 mV: Übergangscharakter. positive Werte: aerobe Vorgänge.")
24
Voltammetrie - Polarographie
Voltametrie Voltammetrie - Polarographie Unter dem Begriff Voltammetrie (Volt-ampero-metrie) versteht man die messtechnische Aufnahme von Strom-Spannungs-Kurven. Gemäß IUPAC Definition beinhaltet der Begriff Voltammetrie das Arbeiten mit stationären oder festen Arbeitselektroden (HMDE, TMFE, GCE, CPE). Erfolgt die Aufnahme der Strom-Spannungs-Kurven dagegen mit flüssigen Arbeitselektroden, deren Oberfläche periodisch oder kontinuierlich erneuert wird, so bezeichnet man diese Messmethode als Polarographie (DME, SMDE) . Eines der Hauptanwendungsgebiete polarogra-phischer und voltammetrischer Verfahren besteht in der Spuren- und Ultraspuren-Bestimmung von Me-tallen in den verschiedensten Umweltkompartimenten, in Getränken und Lebensmitteln.
 versteht man die messtechnische Aufnahme von Strom-Spannungs-Kurven. Gemäß IUPAC Definition beinhaltet der Begriff Voltammetrie das Arbeiten mit stationären oder festen Arbeitselektroden (HMDE, TMFE, GCE, CPE). Erfolgt die Aufnahme der Strom-Spannungs-Kurven dagegen mit flüssigen Arbeitselektroden, deren Oberfläche periodisch oder kontinuierlich erneuert wird, so bezeichnet man diese Messmethode als Polarographie (DME, SMDE) . Eines der Hauptanwendungsgebiete polarogra-phischer und voltammetrischer Verfahren besteht in der Spuren- und Ultraspuren-Bestimmung von Me-tallen in den verschiedensten Umweltkompartimenten, in Getränken und Lebensmitteln.")
25
Voltametrie Messanordnung PC GE AE RE

26
Arbeitsprinzip der Voltametrie/Polarographie
Substanzen werden oxidiert oder reduziert Konzentrationsabnahme im Bereich Elektrodenoberfläche Entstehung eines Konzentrationsgradienten Nachlieferung der Substanz wird durch Diffusion bestimmt Ausbildung eines Diffusionsgrenzstromes

27
Theoretische Grundlagen der Polarographie
Voltametrie Theoretische Grundlagen der Polarographie Im einfachsten Falle beruht das Messprinzip der Polarographie auf die Verfolgung des Stromes (i), der bei linearer Spanungsänderung durch die DME fließt. Fließender Strom i = iF + iC iF- Faradayscher Strom (die Oxidation oder Reduktion des Analyten) iC- Kapazitätsstrom (die Auf- und Entladung der Elektrochemischen Doppelschicht auf der Oberfläche der Arbeitselektrode) Verhältnisse von Faradayschen Strom iF zum Kapazitätsstrom iC in einem Gleichstrompolarogramm E1/2 Ilković-Gleichung:
, der bei linearer Spanungsänderung durch die DME fließt. Fließender Strom i = iF + iC. iF- Faradayscher Strom (die Oxidation oder Reduktion des Analyten) iC- Kapazitätsstrom (die Auf- und Entladung der Elektrochemischen Doppelschicht auf der Oberfläche der Arbeitselektrode) Verhältnisse von Faradayschen Strom iF zum Kapazitätsstrom iC. in einem Gleichstrompolarogramm. E1/2. Ilković-Gleichung:")
28
Voltametrie

29
Voltammetrische Methoden
Klassische Direktstrom - Polarographie mit kontinuierlicher Strommessung (Detektionslimit: 10-4 M)
")
30
Puls-Methoden Zu den Pulsmethoden gehören: Normal-Puls-Polarographie,
Differential-Puls-Polarographie Rechteckwellenpolarographie Die allgemeine Merkmal dieser Methoden besteht, dass die Elektrodenvorgänge in unterschiedlicher Weise mit periodisch wechselnden Rechteckspannungen bei gleich bleibender oder wachsender Amplitude angeregt werden. Dabei kommt es zu der Erscheinung, dass während der Pulszeit der Faradaysche-Strom iF mit t-1/2 und der Kapazitätsstrom iC nach e-kt abklingt. Folglich wird bei einer Messung gegen Ende der Pulszeit tp vorwiegend der Faradaysche Anteil erfasst, da der Kapazitätsstrom zu diesem Zeitpunkt fast vollständig abgeklungen ist.

31
Normal-Puls-Methode (Detektionslimit: bis 10-7M)
Bei der Normal-Puls-Polarographie wird, ausgehend von einem bestimmten Basispotential, nur ein kurzer Spannungspuls zum Ende jedes Tropfens angelegt, dessen Höhe von Tropfen zu Tropfen schrittweise gesteigert wird. Die Strommessung erfolgt nur im hinteren Teil des Spannungspulses.

32
Differential-Puls-Methode (Detektionslimit: bis zu 10-8M)
Das Anregungssignal besteht aus einer treppenförmig anwachsenden Gleichspannung, auf die in periodischer Folge kleine Rechteckpulse von gleich bleibender Spannung gesetzt werden.

33
Stripping Verfahren (Detektionslimit bis zu 10-11M)
Die hohe Empfindlichkeit beruht darauf, dass der Analyt vor seiner Bestimmung an der Arbeitselektrode angereichert wird und durch die Potenzialumkerung wird die angereichten Analytspezies abgelöst. Anreicherung Bestimmung Men+ + ne- + (Hg) Me0(Hg) Durch ASV können alle Metalle bestimmt werden, die unter Bildung von Amalgamen in Quecksilber löslich sind.
 Me0(Hg) Durch ASV können alle Metalle bestimmt werden, die unter Bildung von Amalgamen in Quecksilber löslich sind.")
34
Inverse DPP von Trinkwasser

35
Anwendung der Voltametrie/Polarographie in der Wasseranalytik
DIN Teil 16: Bestimmung von 7 Metallen (Zink, Kupfer, Thallium, Blei, Cadmium, Nickel Cobalt) mittels Voltametrie VOLTAMETRIE: Aufnahme von Strom-Spannungskurven an polarisierten Arbeitselektroden (Messung der Stromstärke bei zeitlich veränderter Spannung) POLAROGRAPHIE: spezielle Methode mit einer flüssigen, tropfenden (d. h. sich verändernden) Quecksilberelektrode als Arbeitselektrode, Oberfläche erneuert sich ständig – damit frei von störenden Substanzen
 mittels Voltametrie. VOLTAMETRIE: Aufnahme von Strom-Spannungskurven an polarisierten Arbeitselektroden (Messung der Stromstärke bei zeitlich veränderter Spannung) POLAROGRAPHIE: spezielle Methode mit einer flüssigen, tropfenden (d. h. sich verändernden) Quecksilberelektrode als Arbeitselektrode, Oberfläche erneuert sich ständig – damit frei von störenden Substanzen.")
36
Anwendung der Voltametrie/Polarographie in der Wasseranalytik
qualitative Aussagen: Art der Substanz ist durch das Halbstufenpotential charakterisiert, Steuerung über Zusammensetzung des Grundelektrolyten (Puffer, Zusätze) quantitative Aussagen: Diffusionsgrenzstrom ist konzentrationsabhängig (Höhe der Stufe) Messung von Peakhöhen nach Integration der Stufen, Kalibrierung notwendig Invers-Voltametrie: (Detektionslimit bis zu 10-11M) Möglichkeit zur Anreicherung, zunächst Abscheidung der Metalle an Hg-Elektrode als Amalgame, anschließend Auflösung durch Potentialumkehr und Messung
 quantitative Aussagen: Diffusionsgrenzstrom ist konzentrationsabhängig (Höhe der Stufe) Messung von Peakhöhen nach Integration der Stufen, Kalibrierung notwendig. Invers-Voltametrie: (Detektionslimit bis zu 10-11M) Möglichkeit zur Anreicherung, zunächst Abscheidung der Metalle an Hg-Elektrode als Amalgame, anschließend Auflösung durch Potentialumkehr und Messung.")
37
Voltametrie Typical Detection Limits
(1 ppt = 1 part per trillion = 1 ng/kg) Antimony Sb(III), Sb(V) 200 ppt Arsenic As(III), As(V) 100 ppt Bismuth Bi 500 ppt Cadmium Cd 50 ppt Chromium Cr(III), Cr(VI) 25 ppt Cobalt Co Copper Cu Iron Fe(II), Fe(III) Lead Pb Mercury Hg Molybdenum Mo Nickel Ni Platinum Pt 0.1 ppt Rhodium Rh Thallium Tl Tungsten W Uranium U Zinc Zn
 Antimony. Sb(III), Sb(V) 200 ppt. Arsenic. As(III), As(V) 100 ppt. Bismuth. Bi. 500 ppt. Cadmium. Cd. 50 ppt. Chromium. Cr(III), Cr(VI) 25 ppt. Cobalt. Co. Copper. Cu. Iron. Fe(II), Fe(III) Lead. Pb. Mercury. Hg. Molybdenum. Mo. Nickel. Ni. Platinum. Pt. 0.1 ppt. Rhodium. Rh. Thallium. Tl. Tungsten. W. Uranium. U. Zinc. Zn.")
38
Amperometrische Sauerstoffbestimmung
Amperometrie: -Spannung Arbeitselektrode konstant -Messung von Diffusionsströmen -Proportionalität zur Analytkonzentration Sauerstoffbestimmung -Reduktion an der Katode O e H 2 H2O (saure Lösung) O e H2O 4 OH (alkalische Lösung) -Katode (inert, z.B. Platin) negativ aufgeladen -Sauerstoff diffundiert durch spezielle Membran, wird reduziert -stufenförmiger Anstieg der Stromstärke, gegen Grenzwert
 O2 + 4 e- + 2 H2O 4 OH- (alkalische Lösung) -Katode (inert, z.B. Platin) negativ aufgeladen. -Sauerstoff diffundiert durch spezielle Membran, wird reduziert. -stufenförmiger Anstieg der Stromstärke, gegen Grenzwert.")
39
-Diffusionsgrenzstrom proportional zur Konzentration Pm
IGR Diffusionsgrenzstrom n Anzahl der Ladungsträger A Elektrodenoberfläche L Dicke der elektrochem. Doppelschicht Pm Permeabilitätskoeffizient Pm IGR = n F A ______ C (O2) L - Membran (z.B. Teflon) verhindert andere Redoxreaktionen - Anströmgeschwindigkeit mind. 0,25 m/s
 L. - Membran (z.B. Teflon) verhindert andere Redoxreaktionen. - Anströmgeschwindigkeit mind. 0,25 m/s.")
40
Amperometrische Bestimmung von freiem Chlor

41
Amperometrische Bestimmung von CSB
bordotierte Diamantelektrode: hohe Ausbeute OH-Bildung geringer Grundstrom chemisch, mechanisch stabill Reaktionsmechanismus: k1 1. Bildung physisorbierter OH-Radikale: BDD[] + H2O BDD[●OH] + H+ + e- k2 Elektrochemische Verbrennung: R + BDD[●OH] BDD[] + CO2 + H2O + ... I = FAk2ZcCSB/8000 F Faraday-Konstante A Elektrodenfläche k2 Geschwindigkeitskonstante Zc Stöchiometriefaktor für vollst. Abbau des Reaktanden

42
Amperometrische Bestimmung von CSB
Bestimmungsgrenze 1 mg/L CSB Analysenzeit 3 Minuten

Ähnliche Präsentationen
 die Lösung ist.>")


















