Präsentation herunterladen
Die Präsentation wird geladen. Bitte warten

1
Hämorrhagische Diathesen L. Braunert, Abteilung für Hämatologie und Internistische Onkologie, Universitätsklinikum Leipzig

2
Hämorrhagische Diathesen Definition: Hämorrhagische Diathese = generalisierte Blutungsneigung

3
Gliederung 1. Physiologie der Blutgerinnung 2. Klinische Diagnostik 3. Labordiagnostik 4. Spezielle hämorrhagische Diathesen 1. Vasopathien 2. Erkrankungen der Thrombozyten 3. Koagulopathien

4
Gliederung 1. Physiologie der Blutgerinnung 2. Klinische Diagnostik 3. Labordiagnostik 4. Spezielle hämorrhagische Diathesen 1. Vasopathien 2. Erkrankungen der Thrombozyten 3. Koagulopathien

5
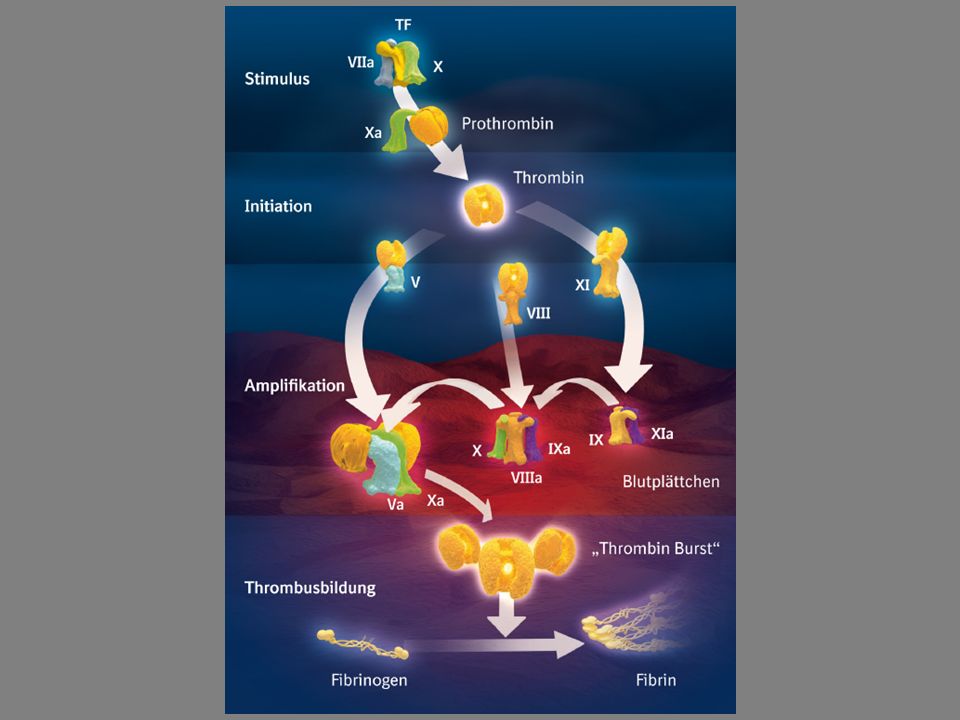
Das Hämostasesystem

6
Thrombozyten Verletzung Gefäßwand ↓ Adhäsion ↓ Formwandel und Freisetzungsreaktion ↓ Aggregation

7
Das plasmatische Gerinnungssystem Bildung des Fibringerinnsels Serinproteasen: II, VII, IX, X, XI, XII Aminotransferase: XIII + Akzeleratoren: V, VIII, HMWK + Phospholipidoberfläche der Thrombozyten

9
Hämorrhagische Diathesen Vasopathie Thrombozytopenie Hämorrhagische Diathese Thrombozytopathie Koagulopathie

10
Gliederung 1. Physiologie der Blutgerinnung 2. Klinische Diagnostik 3. Labordiagnostik 4. Spezielle hämorrhagische Diathesen 1. Vasopathien 2. Erkrankungen der Thrombozyten 3. Koagulopathien

11
Klinische Diagnostik Blutungsanamnese

12
Klinische Diagnostik Untersuchungsbefunde Thrombozytopenie/-pathie Vasopathie Purpura

13
Untersuchungsbefunde Hämatome/ Suggilationen Hämarthros Plasmatische Gerinnungsstörungen Klinische Diagnostik

14
Gliederung 1. Physiologie der Blutgerinnung 2. Klinische Diagnostik 3. Labordiagnostik 4. Spezielle hämorrhagische Diathesen 1. Vasopathien 2. Erkrankungen der Thrombozyten 3. Koagulopathien

15
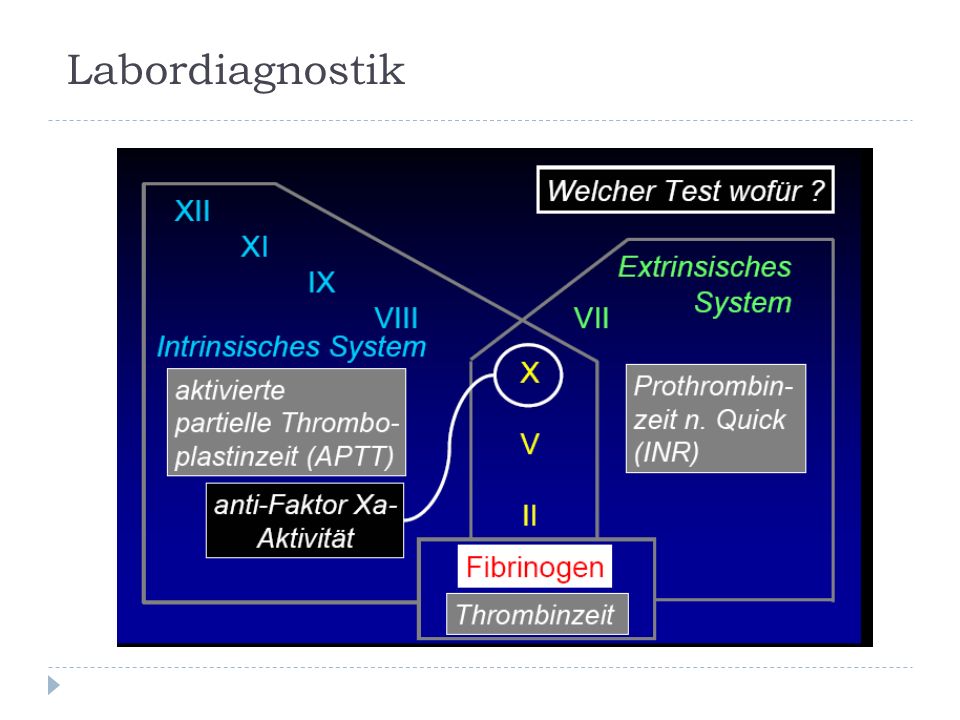
Globaltests: Thrombozytenzählung (150-400 Gpt/l) Aktivierte partielle Thromboplastinzeit (aPTT; Norm: 26-40 sec) Thromboplastinzeit (Quick-Test; Norm: 70-100%) Labordiagnostik
 Aktivierte partielle Thromboplastinzeit (aPTT; Norm: sec) Thromboplastinzeit (Quick-Test; Norm: %) Labordiagnostik")
17
Ergänzung: Blutungszeit (in vivo n. Mielke oder in vitro PFA-100) Thrombinzeit, Reptilasezeit Einzelfaktorenanalyse Analyse des vWF Plättchenfunktionstests und Durchflußzytometrie Labordiagnostik
 Thrombinzeit, Reptilasezeit Einzelfaktorenanalyse Analyse des vWF Plättchenfunktionstests und Durchflußzytometrie Labordiagnostik.")
18
Gliederung 1. Physiologie der Blutgerinnung 2. Klinische Diagnostik 3. Labordiagnostik 4. Spezielle hämorrhagische Diathesen 1. Vasopathien 2. Erkrankungen der Thrombozyten 3. Koagulopathien

19
1. Vasopathien Angeborene Vasopathien M. Osler: hereditäre hämorrhagische Teleangiektasie Erworbene Vasopathien Purpura simplex Purpura senilis Infektiös und medikamentös bedingte Purpura Purpura Schoenlein-Henoch

20
Quantitative Defekte = Thrombozytopenie Klinik:petechiale Blutungen Ursachen:Verminderte Bildung Vermehrte Destruktion/ Sequestration Diagnostik:Blutausstrich Thrombozytenzahl Knochenmarkuntersuchung 2. Erkrankungen der Thrombozyten

21
Immunthrombozytopenie Definition: Abbau mit Autoantikörpern beladener Thrombozyten im RES Klinik: Haut- und Schleimhautblutungen Pathogenese: IgG-AK gegen GP Ib/IX oder GP IIb/IIIa → Bindung an Fc-Rezeptoren der Makrophagen → RES Milz ist Hauptbildungsort der AK und Hauptabbauort der TZ

22
Diagnostik: Ausschluß Pseudothrombopenie, AK-Nachweis, KMP Therapie: - Kortikosteroide - Immunglobuline - Splenektomie - TPO-Rezeptor-Agonisten Immunthrombozytopenie

23
Qualitative Defekte = Thrombozytopathien Klinik: petechiale Blutungen Diagnostik: Blutungszeit Thrombozytenzahl Aggregometrie Durchflußzytometrie 2. Erkrankungen der Thrombozyten

24
Angeboren: Bernard-Soulier-Syndrom (Defekt/Mangel der GP Ib/IX-Komplexe) Thrombasthenie Glanzmann (Defekt/Mangel der GP IIb/IIIa-Komplexe) Abnorme Plättchenfreisetzung Erworben: Systemerkrankungen ASS, Clopidogrel Thrombozytopathien GPIb/V/IX GPIIb/IIIa Rezeptordefekte für lösliche Agonisten (ADP, TXA2) Delta-Storage- Pool-Defekte Bernard Soulier-Syndrom Thrombasthenie Glanzmann
 Thrombasthenie Glanzmann (Defekt/Mangel der GP IIb/IIIa-Komplexe) Abnorme Plättchenfreisetzung Erworben: Systemerkrankungen ASS, Clopidogrel Thrombozytopathien GPIb/V/IX GPIIb/IIIa Rezeptordefekte für lösliche Agonisten (ADP, TXA2) Delta-Storage- Pool-Defekte Bernard Soulier-Syndrom Thrombasthenie Glanzmann")
25
Angeborene plasmatische Gerinnungsstörungen Aktivitätsminderung eines Gerinnungsfaktors mit verzögerter/unzureichender Fibrinbildung Hämophilie Von-Willebrand-Syndrom 3. Koagulopathien

26
Hämophilie Hämophilie A (HA) = FVIII-Mangel Hämophilie B (HB) = FIX-Mangel Verhältnis HA : HB = 5 : 1 Vererbung: X-chromosomal
 = FVIII-Mangel Hämophilie B (HB) = FIX-Mangel Verhältnis HA : HB = 5 : 1 Vererbung: X-chromosomal")
27
Blutungslokalisation: 80% Gelenke 13% Muskulatur (Psoas, Wade) 7% viscerale Blutungen/ ICB Komplikation hämophile Arthropathie! Hämophilie

28
Diagnostik: aPTT verlängert Quick, Blutungszeit normal Einzelfaktorenanalyse mit Restaktivität Molekularbiologie Hämophilie

29
Therapie: Substitution von Faktor VIII/IX 1. Dauerbehandlung 2. Bedarfsbehandlung DDAVP (Minirin®) Hämophilie
 Hämophilie.")
30
FVIIIa FVIIIi APC PS FV FVIII-VWF-Komplex VWF Endothel VWF Flow subendotheliale Matrix Plt Von-Willebrand-Syndrom Quantitativer Mangel oder qualitativer Defekt des von-Willebrand-Faktors

31
Vererbung: autosomal Klinik: Typ 1 und 2 Schleimhautblutungen Typ 3 hämophilieähnliches Bild Nachblutung nach Operation/Zahnextraktion Von-Willebrand-Syndrom Typ 1: Quantitativer DefektVerlaufsform meist leicht Typ 2: Qualitativer Defekt Verlaufsform meist leicht bis mittelschwer Typ 3: Quantitativer DefektVerlaufsform meist schwer

32
Diagnostik: aPTT verlängert Blutungszeit verlängert vWF-Ag vermindert vWF-Aktivität und FVIII-Aktivität erniedrigt Aggregation mit Ristocetin pathologisch Multimeranalyse Von-Willebrand-Syndrom

33
Therapie: DDAVP (Minirin®; Octostim®) vWF haltige Faktor-VIII-Konzentrate (Haemate®) Antifibrinolytika (Tranexamsäure) Von-Willebrand-Syndrom
 vWF haltige Faktor-VIII-Konzentrate (Haemate®) Antifibrinolytika (Tranexamsäure) Von-Willebrand-Syndrom")
34
3. Koagulopathien Erworbene plasmatische Gerinnungsstörungen Vorkommen bei einer Vielzahl von Erkrankungen Mehrere Gerinnungsfaktoren betroffen Vitamin-K-Mangelzustände Lebererkrankungen Disseminierte intravasale Gerinnung (DIC) Erworbene Inhibitoren
 Erworbene Inhibitoren.")
35
3. Koagulopathien Hyperfibrinolyse Überschießender Fibrinabbau Diagnostik: Thrombelastogramm Fibrinogen vermindert D-Dimer erhöht Therapie: Antifibrinolytika (Tranexamsäure)
.")
36
Vielen Dank für Ihre Aufmerksamkeit!

Ähnliche Präsentationen
















, Oliver Dollase (EvKB)>")


