Präsentation herunterladen
Die Präsentation wird geladen. Bitte warten

1
Diabetesassistentinnen-Ausbildung
Willkommen zur Diabetesassistentinnen-Ausbildung CEB Fortbildungswerk, Merzig-Hilbringen Frühjahr 2007 „Pathophysiologie – Metabolisches Syndrom“ Dr. med. Armin Sammler Internist – Gastroenterologe – Diabetologe

2
Blutglukoseregulation und das Auftreten eines Typ 2 Diabetes
In dieser Information werden die Blutglukoseregulation und die Faktoren diskutiert, die zum Verlust der Blutglukoseregulation und zum Auftreten eines Typ 2 Diabetes führen.

3
Die weltweite Epidemie des Typ 2 Diabetes:
Betazellfunktionsstörung und Insulinresistenz Im Folgenden werden die Zusammenhänge zwischen verminderter Betazellfunktion, erhöhter Insulinresistenz und Auftreten eines Typ 2 Diabetes untersucht.

4
Globale Hochrechnungen für die Diabetesepidemie: 2003–2025 (in Millionen)
48,4 58,6 23,0 36,2 39,3 81,6 19,2 39,4 DISKUSSION Die größten Anstiege der Prävalenz werden für Indien, China und die Vereinigten Staaten vorhergesagt. Weltweit gibt es vermutlich über 300 Millionen Menschen mit gestörter Glukosetoleranz. Bei den Schätzungen handelt es sich um Hochrechnungen aus 7 Regionen der International Diabetes Federation, die unten stehend aufgeführt sind: Region Afrika 7,1 15,0 Östliche Mittelmeerregion und Mittlerer Osten 19,2 39,4 Europa 48,4 58,6 Nordamerika 23,0 36,2 Süd- und Mittelamerika 14,2 26,2 Südostasien 39,3 81,6 Westlicher Pazifik 43,0 75,8 7,1 15,0 43,0 75,8 14,2 26,2 2003 = 194 Millionen 2025 = 333 Millionen Anstieg um 72 % Sicree R, et al. In: Gan D, Hrsg. Diabetes Atlas. 2. Ausgabe. Brüssel: International Diabetes Federation; 2003:15-71 Zimmet P. AM J Med. 2005;118(Suppl 2):3S-8S.
:3S-8S.")
5
Geschätzte Prävalenz des Diabetes in der
globalen Erwachsenenpopulation, 350 300 250 200 150 100 50 300 228 DISKUSSION Es wurden Schätzungen zum Ausmaß der Diabetesepidemie in den Industrieländern und den Schwellenländern durchgeführt. Die stärkste Auswirkung der Diabetesepidemie wird in den Schwellenländern erwartet. Millionen 135 84 72 51 Weltweit Industrieländer Schwellenländer King H, et al. Diabetes Care. 1998;21: Nachdruck mit Erlaubnis der American Diabetes Association.

6
Die zugrunde liegenden Schädigungen: Insulinresistenz und Betazellfunktionsstörung
Bewegungs-armer Lebensstil Gluko-toxizität Genetische Prädispositionen ↑ freie Fettsäuren Ernährung Adipositas Insulinresistenz (IR) Betazellfunktionsstörung Gestörte Insulinantwort Gestörte Insulinproduktion und -sekretion - Hyperinsulinämie - Normale Glukosetoleranz DISKUSSION Das Auftreten eines Typ 2 Diabetes hat Schädigungen in zahlreichen Organsystemen zur Folge, die durch eine Vielzahl von Faktoren verschlimmert werden können, zum Beispiel Ernährung und Lebensstil, genetische Ursachen und Fehlregulation des Fett- und Kohlenhydratstoffwechsels. Am Anfang ist die Insulinresistenz mit Hyperinsulinämie und einer normalen Glukosetoleranz verbunden. Die Betazellfunktionsstörung äußert sich in Form einer gestörten Insulinproduktion und einer unzureichenden Sekretion von Insulin zur Kompensation der Insulinresistenz. Schließlich übersteigt der Insulinbedarf die Kapazität der Betazellen; dies führt zu einer gestörten Glukosetoleranz, einer zunehmenden Hyperglykämie und zum Übergang in einen Typ 2 Diabetes. Versagen der Betazellen, die IR zu kompensieren IR + sinkende Insulinspiegel + gestörte Glukosetoleranz Typ 2 Diabetes Saltiel AR, et al. Diabetes. 1996;45: ; Ramlo-Halsted BA, et al. Prim Care. 1999;26:
 Betazellfunktionsstörung. Gestörte. Insulinantwort. Gestörte Insulinproduktion. und -sekretion. - Hyperinsulinämie. - Normale Glukosetoleranz. DISKUSSION. Das Auftreten eines Typ 2 Diabetes hat Schädigungen in zahlreichen Organsystemen zur Folge, die durch eine Vielzahl von Faktoren verschlimmert werden können, zum Beispiel Ernährung und Lebensstil, genetische Ursachen und Fehlregulation des Fett- und Kohlenhydratstoffwechsels. Am Anfang ist die Insulinresistenz mit Hyperinsulinämie und einer normalen Glukosetoleranz verbunden. Die Betazellfunktionsstörung äußert sich in Form einer gestörten Insulinproduktion und einer unzureichenden Sekretion von Insulin zur Kompensation der Insulinresistenz. Schließlich übersteigt der Insulinbedarf die Kapazität der Betazellen; dies führt zu einer gestörten Glukosetoleranz, einer zunehmenden Hyperglykämie und zum Übergang in einen Typ 2 Diabetes. Versagen der Betazellen, die IR zu kompensieren. IR + sinkende Insulinspiegel + gestörte Glukosetoleranz. Typ 2 Diabetes. Saltiel AR, et al. Diabetes. 1996;45: ; Ramlo-Halsted BA, et al. Prim Care. 1999;26:")
7
PostprandialeBlutglukose Diagnosestellung des Diabetes
Natürlicher Verlauf des Typ 2 Diabetes: Insulinresistenz und Betazellfunktionsstörung Gestörte Glukosetoleranz Diabetes Insulinsekretion Betazellfunktionsstörung Normales Insulin Insulinresistenz Hyperinsulinämie Leichte postprandiale Hyperglykämie Betazellversagen Postprandiale Hyperglykämie Nüchternhyperglykämie DISKUSSION Diese Abbildung zeigt den Übergang einer gestörten Glukosetoleranz in einen Typ 2 Diabetes. Metabolische Abweichungen, die die gestörte Glukosetoleranz kennzeichnen, sind mindestens 4–7 Jahre vorhanden, bevor sie manifest werden, und sie gehen der klinischen Diagnose des Diabetes um 9–12 Jahre voraus. Die sich verschlechternde Betazellfunktion und der darauf folgende Verlust der Insulinreaktion führen zu einer erhöhten postprandialen Blutglukose. Die Nüchternblutglukose bleibt während der Anfangsstadien dieses Zyklus normal und steigt mit fortschreitendem Typ 2 Diabetes an. Der Typ 2 Diabetes ist durch eine anhaltende Insulinresistenz, einen Insulinmangel und einen Betazellverlust gekennzeichnet. PostprandialeBlutglukose Normale Blutglukose Nüchternblutglukose Diagnosestellung des Diabetes 4–7 Jahre Modifiziert nach Ramlo-Halsted BA, et al. Prim Care. 1999;26: Reprinted from Primary Care, 26, Ramlo-Halsted BA, et al. The natural history of type 2 diabetes, , Copyright 1999, mit Erlaubnis von Elsevier.

8
Die Blutglukosekontrolle: Ein komplexer Prozess
Im Folgenden werden die grundlegenden Aspekte der Regulation der Blutglukosespiegel durch den Körper nach Nahrungsaufnahme dargestellt.

9
Insulin und Glukagon steuern die normale postprandiale Blutglukoseregulation
140 Mahlzeit Blutglukose Insulin 120 Glukagon mg% 100 Nach einer Mahlzeit 160 120 mU/ml 80 DISKUSSION Die Insulinfreisetzung steigt als Reaktion auf eine Mahlzeit an, während die Glukagonspiegel supprimiert werden. Diese Trends normalisieren sich, wenn die Blutglukosespiegel wieder auf die Nüchternwerte sinken (ca. 2 Stunden nach einer Mahlzeit). Bei gesunden Probanden wird die Blutglukose nach einer Mahlzeit in einem relativ schmalen Bereich gehalten (ca. 20–40 mg/dl). HINTERGRUND Plasmainsulin und -glukagon wurden bei gesunden Probanden ohne Diabetes nach einer Mahlzeit gemessen. Insulin Glukagon 40 130 Dieses Zusammenspiel reduziert Blutglukose-anstiege nach einer Mahlzeit auf ein Minimum 120 110 pg/ml 100 90 -60 60 120 180 240 Zeit (min) Unger RH. N Eng J Med. 1971;285: Copyright © 1971 Massachusetts Medical Society. Alle Rechte vorbehalten. übersetzt mit Erlaubnis 2006.
. Bei gesunden Probanden wird die Blutglukose nach einer Mahlzeit in einem relativ schmalen Bereich gehalten (ca. 20–40 mg/dl). HINTERGRUND. Plasmainsulin und -glukagon wurden bei gesunden Probanden ohne Diabetes nach einer Mahlzeit gemessen. Insulin Glukagon Dieses Zusammenspiel reduziert Blutglukose-anstiege nach einer Mahlzeit auf ein Minimum pg/ml Zeit (min) Unger RH. N Eng J Med. 1971;285: Copyright © 1971 Massachusetts Medical Society. Alle Rechte vorbehalten. übersetzt mit Erlaubnis")
10
Die Blutglukoseregulation ist ein Prozess, an dem mehrere Organe beteiligt sind
Zentrales Nervensystem Nahrungsaufnahme & Sättigung Hormonregulation Periphere Zielgewebe Muskel: Glukoseaufnahme und -Verwertung Leber: Glukoneogenese Pankreas Betazellen: Insulinsekretion Alphazellen: Glukagonsekretion Verdauungssystem Glukoseresorption Inkretinhormone Flint A, et al. J Clin Invest ;101: ; Unger RH. N Eng J Med. 1971;285: ; Mitrakou A, et al. Diabetes. 1990;39: ; Saltiel AR, et al. Diabetes. 1996;45:

11
Das endokrine Pankreas steuert die Blutglukosehomöostase
Überwacht ständig die Blutglukosekonzentration Produziert Hormone, die den Blutglukosespiegel verändern Insulin Glukagon Steuert die Hormonfreisetzung zur Aufrechterhaltung der Blutglukosehomöostase HINTERGRUND Im Inselorgan des Pankreas wird Insulin von den Betazellen und Glukagon von den Alphazellen gebildet.

12
Plasmainsulin (U/ml)
Die gestörte Insulinantwort ist mit einer Nüchternhyperglykämie verbunden Nüchternhyperglykämie > 150 mg/dl Nüchternhyperglykämie 110–150 mg/dl Normale Blutglukose 500 120 100 400 DISKUSSION Patienten mit Nüchternblutglukosespiegeln > 150 mg/dl hatten nach oraler Glukosebelastung einen minimalen Anstieg des mittleren Plasmainsulinspiegels. Bei Patienten mit Nüchternblutglukosespiegeln im Bereich von 110–150 mg/dl zeigt sich dabei ein der Insulinspiegel; dieser war jedoch nicht schnell genug, um den erheblichen Anstieg der postprandialen Blutglukosekonzentration zu verhindern. Bei Patienten mit normalem Nüchternblutglukosespiegel kam es nach oraler Blutglukosebelastung zu einem maximalen Anstieg des Plasmainsulins. Die Nüchterninsulinspiegel waren in den drei für die Blutglukoseregulation repräsentativen Gruppen identisch. HINTERGRUND Die Studien-Stichprobe umfasste 41 Patienten (n = 21 mit normaler Nüchternblutglukose, n=9 mit Nüchternhyperglykämie von 100– 150 mg/dl, n = 11 mit Nüchternhyperglykämie > 150 mg/dl). Die Patienten waren hinsichtlich Alter, Geschlechtsverteilung und Gewicht angemessen gematcht. Die Patienten konsumierten über 3 Tage 3-mal täglich eine Standard-Flüssigdiät. Nach 3 Tagen erhielten die Patienten eine orale Glukosebelastung von 40 g/m2 Körperoberfläche; anschließend wurden 3 Stunden lang Blutproben entnommen. Die Patienten waren vorher nie mit Insulin behandelt worden. Zwei Studienuntergruppen, die zwischen normaler Blutglukose und der Nüchternhyperglykämie von 110–150 mg/dl lagen, sind hier nicht abgebildet. 80 300 Blutglukose (mg%) Plasmainsulin (U/ml) 60 200 40 100 20 0,0 0,5 1,0 2,0 3,0 0,0 0,5 1,0 2,0 3,0 Zeit (Stunden) Zeit (Stunden) n=41 Reaven G, et al. Am J Med. 1976;60:80-88. Nachdruck aus American Journal of Medicine. Vol 60, Reaven G, et al, Copyright 1976, mit Erlaubnis von Excerpta Medica Inc.
. Die Patienten waren hinsichtlich Alter, Geschlechtsverteilung und Gewicht angemessen gematcht. Die Patienten konsumierten über 3 Tage 3-mal täglich eine Standard-Flüssigdiät. Nach 3 Tagen erhielten die Patienten eine orale Glukosebelastung von 40 g/m2 Körperoberfläche; anschließend wurden 3 Stunden lang Blutproben entnommen. Die Patienten waren vorher nie mit Insulin behandelt worden. Zwei Studienuntergruppen, die zwischen normaler Blutglukose und der Nüchternhyperglykämie von 110–150 mg/dl lagen, sind hier nicht abgebildet Blutglukose (mg%) Plasmainsulin (U/ml) ,0. 0,5. 1,0. 2,0. 3,0. 0,0. 0,5. 1,0. 2,0. 3,0. Zeit (Stunden) Zeit (Stunden) n=41. Reaven G, et al. Am J Med. 1976;60: Nachdruck aus American Journal of Medicine. Vol 60, Reaven G, et al, Copyright 1976, mit Erlaubnis von Excerpta Medica Inc.")
13
Patienten mit Typ 2 Diabetes haben höhere Nüchtern-blutglukosespiegel und höhere postprandiale Blutglukosespiegel Mahlzeit I Mahlzeit II Mahlzeit III Kontrollprobanden Patienten mit Typ 2 Diabetes 400 300 DISKUSSION Die Nüchternblutglukosespiegel und die postprandialen Blutglukosespiegel waren bei Patienten mit Typ 2 Diabetes wesentlich höher als bei Probanden ohne Diabetes. HINTERGRUND Dieses Dia zeigt Blutglukosekurven, die in einer Studie an 16 Patienten mit unbehandeltem Typ 2 Diabetes und 14 Kontrollprobanden (nach Alter, Geschlecht und Ausmaß der Adipositas gematcht) gemessen wurden. Alle Teilnehmer erhielten während eines Zeitraums von 24 Stunden 3 gemischte Mahlzeiten. Die Nüchternblutglukosespiegel und die postprandialen Blut- zuckerspiegel waren bei Patienten mit Typ 2 Diabetes signifikant höher p < 0,0001 Blutglukose (mg/dl) 200 100 600 1.000 1.400 1.800 2.200 2.600 3.000 Zeit (Stunden) Polonsky KS, et al. N Engl J Med. 1988;318: Copyright © 1998 Massachusetts Medical Society. Alle Rechte vorbehalten. übersetzt mit Erlaubnis 2006.
 gemessen wurden. Alle Teilnehmer erhielten während eines Zeitraums von 24 Stunden 3 gemischte Mahlzeiten. Die Nüchternblutglukosespiegel und die postprandialen Blut- zuckerspiegel waren bei Patienten mit Typ 2 Diabetes signifikant höher p < 0,0001. Blutglukose (mg/dl) Zeit (Stunden) Polonsky KS, et al. N Engl J Med. 1988;318: Copyright © 1998 Massachusetts Medical Society. Alle Rechte vorbehalten. übersetzt mit Erlaubnis")
14
Die postprandiale Hyperglykämie geht einher mit einer gestörten Insulinsekretion und Glukagonsuppression Orale Glukose 20 Kontrollprobanden Patienten mit Typ 2 Diabetes 15 Blutglukose (mmol/l) 10 5 DISKUSSION Die Blutglukosewerte bei Patienten mit Typ 2 Diabetes lagen postprandial und im Nüchternzustand im hyperglykämischen Bereich. Bei Patienten mit Typ 2 Diabetes werden maximale postprandiale Blutglukosespiegel später erreicht, und es dauert länger, bis diese wieder den Ausgangszustand erreichen. HINTERGRUND Die 3 auf diesem Dia dargestellten Kurven stammen aus einer Studie an 10 Patienten mit Typ 2 Diabetes und 10 gematchten Kontrollprobanden, die eine orale Glukosebelastung erhielten. Orale Glukose -60 60 120 180 240 300 420 Zeit (min) Orale Glukose 360 75 300 60 Insulin (p mol/l) 240 Glukagon (f mol/l) 180 45 120 30 60 15 -60 60 120 180 240 300 -60 60 120 180 240 300 Zeit (min) Zeit (min) Mitrakou A, et al. Diabetes. 1990;39: Copyright © 1990 American Diabetes Association. Aus Diabetes, Vol 39, 1990; Nachdruck mit Erlaubnis der American Diabetes Association.
 DISKUSSION. Die Blutglukosewerte bei Patienten mit Typ 2 Diabetes lagen postprandial und im Nüchternzustand im hyperglykämischen Bereich. Bei Patienten mit Typ 2 Diabetes werden maximale postprandiale Blutglukosespiegel später erreicht, und es dauert länger, bis diese wieder den Ausgangszustand erreichen. HINTERGRUND. Die 3 auf diesem Dia dargestellten Kurven stammen aus einer Studie an 10 Patienten mit Typ 2 Diabetes und 10 gematchten Kontrollprobanden, die eine orale Glukosebelastung erhielten. Orale Glukose Zeit (min) Orale Glukose Insulin (p mol/l) 240. Glukagon (f mol/l) Zeit (min) Zeit (min) Mitrakou A, et al. Diabetes. 1990;39: Copyright © 1990 American Diabetes Association. Aus Diabetes, Vol 39, 1990; Nachdruck mit Erlaubnis der American Diabetes Association.")
15
Gestörte erste Phase der Insulinsekretion bei Typ 2 Diabetes
Kontrollprobanden Patienten mit Typ 2 Diabetes iv-Glukose 120 100 100 80 80 DISKUSSION Im Gegensatz zur normalen Insulinfreisetzung als Reaktion auf intravenöse Glukosegabe findet sich bei Patienten mit Typ 2 Diabetes praktisch keine erste Phase der Insulinsekretion. HINTERGRUND Auf diesem Dia ist die akute Insulinsekretion auf intravenöse Glukosegabe (20 g) bei gewichts- und geschlechtsgematchten Personen (mit Diabetes, n = 15; ohne Diabetes, n = 18) dargestellt. 60 Insulinfreisetzung (U/ml) Insulinfreisetzung (U/ml) iv-Glukose 60 40 40 20 20 -40 -30 -20 -10 10 20 30 -40 -30 -20 -10 10 20 30 Zeit (min) Zeit (min) Porte D. Diabetes. 1991;40; Copyright © 1991 American Diabetes Association. Aus Diabetes, Vol 40, 1991; Nachdruck mit Erlaubnis der American Diabetes Association.
 bei gewichts- und geschlechtsgematchten Personen (mit Diabetes, n = 15; ohne Diabetes, n = 18) dargestellt. 60. Insulinfreisetzung (U/ml) Insulinfreisetzung (U/ml) iv-Glukose Zeit (min) Zeit (min) Porte D. Diabetes. 1991;40; Copyright © 1991 American Diabetes Association. Aus Diabetes, Vol 40, 1991; Nachdruck mit Erlaubnis der American Diabetes Association.")
16
Der natürliche Verlauf des Typ 2 Diabetes:
Eine fortschreitende Erkrankung Im Folgenden wird der natürliche Verlauf des Typ 2 Diabetes, einer fortschreitenden Krankheit, dargestellt.

17
Der Typ 2 Diabetes ist eine fortschreitende Erkrankung: 6-Jahresdaten der UKPDS
Konventionell Chlorpropamid Glibenclamid Insulin Metformin 9 8 DISKUSSION Der mediane HbA1c stieg im Zeitraum von 6 Jahren nach der Randomisierung in allen in die sekundäre UKPDS-Analyse eingeschlossenen Behandlungsgruppen an. Bei den mit Glibenclamid, Metformin, Chlorpropamid oder Insulin behandelten Patienten kam es im ersten Jahr nach der Randomisierung zu einem Abfall des HbA1c. HINTERGRUND Die UKPDS beobachtete 753 Patienten mit neu diagnostiziertem Typ 2 Diabetes über 10 Jahre in einer randomisierten kontrollierten Studie mit konventioneller Therapie (vorwiegend Diät) im Vergleich zu intensiver Blutglukoseeinstellung mit Metformin (Zielwert für die Nüchternblutglukose unter 6 mmol/l). Eine sekundäre Analyse verglich 342 Patienten, die Metformin zugeordnet waren, mit 951 übergewichtigen Patienten, die einer intensiven Blutglukoseeinstellung mit Chlorpropamid (n = 265), Glibenclamid (n = 277) oder Insulin (n = 409) zugeordnet waren. Der mediane HbA1c während des 10-jährigen Follow-ups betrug in der Metformingruppe 7,4 % und in der Gruppe mit konventioneller Behandlung 8,0 %. Die Patienten, die einer intensiven Blutglukoseeinstellung mit einem Sulfonylharnstoff oder Insulin zugeordnet waren, hatten einen ähnlichen HbA1c wie in der Metformingruppe. HbA1c (Median %) 7 6,2% HbA1c Obergrenze des Normalbereichs 6 1 2 3 4 5 6 Zeit ab der Randomisierung (Jahre) UKPDS. Lancet. 1998;352: Reprinted from Lancet, 352, UKPDS, Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34), Copyright 1998, mit Erlaubnis von Elsevier.
 im Vergleich zu intensiver Blutglukoseeinstellung mit Metformin (Zielwert für die Nüchternblutglukose unter 6 mmol/l). Eine sekundäre Analyse verglich 342 Patienten, die Metformin zugeordnet waren, mit 951 übergewichtigen Patienten, die einer intensiven Blutglukoseeinstellung mit Chlorpropamid (n = 265), Glibenclamid (n = 277) oder Insulin (n = 409) zugeordnet waren. Der mediane HbA1c während des 10-jährigen Follow-ups betrug in der Metformingruppe 7,4 % und in der Gruppe mit konventioneller Behandlung 8,0 %. Die Patienten, die einer intensiven Blutglukoseeinstellung mit einem Sulfonylharnstoff oder Insulin zugeordnet waren, hatten einen ähnlichen HbA1c wie in der Metformingruppe. HbA1c (Median %) 7. 6,2% HbA1c Obergrenze des Normalbereichs Zeit ab der Randomisierung (Jahre) UKPDS. Lancet. 1998;352: Reprinted from Lancet, 352, UKPDS, Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34), Copyright 1998, mit Erlaubnis von Elsevier.")
18
Die Betazellfunktion sinkt im Zeitverlauf stetig um etwa 6 % pro Jahr
Die Betazellfunktion nimmt bei Patienten mit Typ 2 Diabetes im Zeitverlauf ab 100 80 Die Betazellfunktion kann zum Zeitpunkt der Diagnose bereits um 50 % reduziert sein 60 DISKUSSION Die Daten sprechen dafür, dass die Betazellfunktionsstörung bereits Jahre vor dem Auftreten der Hyperglykämie beginnt. Die Betazellfunktion kann zum Zeitpunkt der Diagnosestellung des Diabetes bereits um 50 % gesunken sein. Das Ausmaß der verbleibenden Betazellfunktion ist von entscheidender Bedeutung, da die Betazellfunktion nach der Diagnosestellung mit einer Rate von ungefähr 6 % pro Jahr weiter sinkt. HINTERGRUND Die Abbildung beruht auf dem 6-jährigen Follow-up der UKPDS. Die Betazellfunktion nahm trotz Diät oder Behandlung mit einem Sulfonylharnstoff oder Metformin stetig ab. Betazellfunktion (%) 40 Die Betazellfunktion sinkt im Zeitverlauf stetig um etwa 6 % pro Jahr 20 -12 -10 -8 -6 -4 -2 2 4 6 Jahre ab der Diagnose Holman RR. Diabetes Res Clin Pract. 1998;40(Suppl 1):S21-S25.; UKPDS. Diabetes. 1995;44: Nachdruck aus Diabetes Research and Clinical Practice, 40, Holman RR, Analysis of the United Kingdom Prospective Diabetes Study, S21-S25, Copyright 1998, mit Erlaubnis von Elsevier.
 40. Die Betazellfunktion sinkt im Zeitverlauf stetig um etwa 6 % pro Jahr Jahre ab der Diagnose. Holman RR. Diabetes Res Clin Pract. 1998;40(Suppl 1):S21-S25.; UKPDS. Diabetes. 1995;44: Nachdruck aus Diabetes Research and Clinical Practice, 40, Holman RR, Analysis of the United Kingdom Prospective Diabetes Study, S21-S25, Copyright 1998, mit Erlaubnis von Elsevier.")
19
Korrelation zwischen Betazellfunktion und Insulinsensibilität
„Das hyperbolische Gesetz der Blutglukose“ Normale Glukosetoleranz Gestörte Glukosetoleranz Diabetes Betazellfunktion DISKUSSION Bei den Patienten kommt es in dem Maße zu einer gestörten Glukosetoleranz, wie sie die Fähigkeit verlieren, kompensatorische Insulinmengen als Reaktion auf eine verminderte Empfindlichkeit freizusetzen (Abflachung der Kurve). Patienten mit Typ 2 Diabetes weisen bei Änderungen der Insulinempfindlichkeit ein zunehmendes Betazellversagen auf. HINTERGRUND Die Betazellfunktion und die Insulinempfindlichkeit wurden anhand der oralen Glukosetoleranztestung geschätzt. Die für die Erstellung der Abbildung verwendete Stichprobe umfasste: graue Quadrate, normale Glukosetoleranz (n = 483); weiße Kreise, gestörte Glukosetoleranz (n = 71); schwarze Rauten, Typ 2 Diabetes (n = 65). Insulinempfindlichkeit Stumvoll M. Diabetologia. 2004;47: Nachdruck mit Erlaubnis des Springer-Verlags.
. Patienten mit Typ 2 Diabetes weisen bei Änderungen der Insulinempfindlichkeit ein zunehmendes Betazellversagen auf. HINTERGRUND. Die Betazellfunktion und die Insulinempfindlichkeit wurden anhand der oralen Glukosetoleranztestung geschätzt. Die für die Erstellung der Abbildung verwendete Stichprobe umfasste: graue Quadrate, normale Glukosetoleranz (n = 483); weiße Kreise, gestörte Glukosetoleranz (n = 71); schwarze Rauten, Typ 2 Diabetes (n = 65). Insulinempfindlichkeit. Stumvoll M. Diabetologia. 2004;47: Nachdruck mit Erlaubnis des Springer-Verlags.")
20
Die Aufrechterhaltung der Betazellmasse ist ein dynamischer Prozess
Insulinpositive Zellen (Betazellen) Dynamisches Gleichgewicht des Betazellverlusts vs. Betazell-neogenese Mit neuen Betazellen aufgefüllte Inseln Replikation: Epitheliale Gangzellen Proliferation: Stamm- und Präkursorzellen Pankreasgang DISKUSSION Im gesunden Pankreas besteht ein dynamisches Gleichgewicht zwischen Betazellverlust (durch Prozesse wie Apoptose) und Betazellneogenese. Neue insulinproduzierende Betazellen werden durch Replikation aus epithelialen Gallengangszellen gebildet, wie im histologischen Schnitt dargestellt wird. Neue Betazellen werden auch durch Proliferation aus Stamm- und Präkursorzellen gebildet. Butler AE et al. Diabetes. 2003;52: ; Bonner-Weir S. J Mol Endocrinol. 2000;24: ; Finegood DT, et al. Diabetes. 1995;44: Copyright © 2003 American Diabetes Association. Aus Diabetes, Vol 52, 2003; Nachdruck mit Erlaubnis der American Diabetes Association.
 Dynamisches Gleichgewicht des Betazellverlusts vs. Betazell-neogenese. Mit neuen Betazellen aufgefüllte Inseln. Replikation: Epitheliale Gangzellen. Proliferation: Stamm- und Präkursorzellen. Pankreasgang. DISKUSSION. Im gesunden Pankreas besteht ein dynamisches Gleichgewicht zwischen Betazellverlust (durch Prozesse wie Apoptose) und Betazellneogenese. Neue insulinproduzierende Betazellen werden durch Replikation aus epithelialen Gallengangszellen gebildet, wie im histologischen Schnitt dargestellt wird. Neue Betazellen werden auch durch Proliferation aus Stamm- und Präkursorzellen gebildet. Butler AE et al. Diabetes. 2003;52: ; Bonner-Weir S. J Mol Endocrinol. 2000;24: ; Finegood DT, et al. Diabetes. 1995;44: Copyright © 2003 American Diabetes Association. Aus Diabetes, Vol 52, 2003; Nachdruck mit Erlaubnis der American Diabetes Association.")
21
Die Hyperglykämie entsteht, wenn die Betazellarbeitslast zunimmt und die Betazellreaktion abnimmt
↑ Insulinresistenz - Adipositas - Fehlende körperliche Bewegung ↑ Nahrungsaufnahme ↑ Nahrungsresorptionsrate ↑ Glukagonsekretion - ↑ Glukosefreisetzung aus der Leber ↓ Insulinsekretion als Reaktion auf erhöhten Blutglukose ↓ Erste Phase der Insulinreaktion + DISKUSSION Die Faktoren, die zur erhöhten Betazellarbeitslast und zu verringerten Betazellreaktion beitragen, führen gemeinsam zum Auftreten der Hyperglykämie. Die Hyperglykämie ist eingetreten, da der Insulinbedarf die Kapazität überstiegen hat. Hyperglykämie Seely B, et al. In: Moller DE, Hrsg. Insulinresistenz. Wiley; 1993: ; Weyer C, et al. J Clin Invest. 1999;104:

22
Die Betazellfunktion sinkt im Zeitverlauf unter Monotherapie oder unter alleiniger Diät
Sulfonylharnstoffe Diät Metformin 100 80 Die Abnahme der Betazellfunktion kann mit dem Versagen dieser Therapien zusammenhängen, eine strenge Blutglukose-einstellung zu erreichen DISKUSSION Bei den mit Sulfonylharnstoffen oder Metformin behandelten Patienten kam es während des ersten Studienjahres zu einem nachweisbaren Anstieg der normalen Betazellfunktion; während der darauf folgenden 5 Jahre nahm die Betazellfunktion dagegen stetig ab. Bei den ausschließlich mit Diät behandelten Patienten kam es im 6-jährigen Studienzeitraum zu einer stetigen Abnahme der Betazellfunktion. Die Abnahmerate war nach dem 1. Jahr in allen Patientengruppen ähnlich. Die Ergebnisse der UKPDS sprechen dafür, dass das Versagen von Diät allein oder der Monotherapie, eine strenge Blutglukoseeinstellung zu erreichen, mit einer stetigen Abnahme der Betazellfunktion bei dieser Krankheit verbunden ist. HINTERGRUND Eine Substudie der UKPDS untersuchte die Betazellfunktion bei Probanden mit neu diagnostiziertem Typ 2 Diabetes. Die Betazellfunktion wurde mittels Homeostasis Model Assessment (HOMA) untersucht. Die Probanden in dieser Substudie wurden entweder ausschließlich mit Diät (n = 110), Metformin (n = 159) oder einem Sulfonylharnstoff (n = 511) behandelt. Betazellfunktion (%) 60 40 20 1 2 3 4 5 6 Jahre UKPDS. Diabetes. 1995;44: ; Turner RC, et al. JAMA. 1998;281: Copyright © 1995 American Diabetes Association. Aus Diabetes, Vol 44, 1995; Nachdruck mit Erlaubnis der American Diabetes Association.
 untersucht. Die Probanden in dieser Substudie wurden entweder ausschließlich mit Diät (n = 110), Metformin (n = 159) oder einem Sulfonylharnstoff (n = 511) behandelt. Betazellfunktion (%) Jahre. UKPDS. Diabetes. 1995;44: ; Turner RC, et al. JAMA. 1998;281: Copyright © 1995 American Diabetes Association. Aus Diabetes, Vol 44, 1995; Nachdruck mit Erlaubnis der American Diabetes Association.")
23
Gegenwärtige Medikamente zur Behandlung des
Typ 2 Diabetes Im Folgenden werden die Merkmale der Medikamente diskutiert, die gegenwärtig zur Behandlung des Typ 2 Diabetes zur Verfügung stehen.

24
Pharmakologische Therapien bei Typ 2 Diabetes
Beeinflussen die Insulinproduktion (Pankreas) Insulinsekretagoga: Sulfonylharnstoffe und Glinide Ersetzen Insulin (Leber, Muskeln und Fettgewebe) Insulin, Insulinmischungen und Insulinanaloga Beeinflussen die Insulinwirkung Hemmen die hepatische Glukose-Freisetzung (Leber) Biguanide ↓ Insulinresistenz (Leber, Muskeln und Fettgewebe) -Thiazolidinedione und Biguanide Verlangsamen die Kohlenhydratresorption (Darm) Alphaglucosidase-Inhibitoren DISKUSSION Sulfonylharnstoffe und Glinide blockieren den Sulfonylharnstoffrezeptor und die ATP-empfindlichen Kaliumkanäle in den pankreatischen Betazellen. Ihre Hauptnebenwirkungen sind Hypoglykämie und Gewichtszunahme. Insulin entfaltet seine Wirkung über den Insulin-Rezeptor; seine Hauptnebenwirkungen sind Hypoglykämie und Gewichtszunahme. Der Wirkmechanismus der Biguanide (Metformin) ist unbekannt. Ihre wichtigsten Nebenwirkungen sind gastrointestinale Beschwerden und Laktatazidose. Die Thiazolidinedione sind PPAR-gamma-Agonisten. Ihre Hauptnebenwirkungen sind Gewichtszunahme, Ödem und Anämie. Die Alphaglucosidase-Inhibitoren wirken im Darm; ihre wichtigsten Nebenwirkungen sind gastrointestinale Beschwerden. Moller DE. Nature. 2001;414: ; Pickup JC, Williams G, eds. Textbook of Diabetes 2. Malden, MA: Blackwell;2003:45.5.
 Insulinsekretagoga: Sulfonylharnstoffe und Glinide. Ersetzen Insulin (Leber, Muskeln und Fettgewebe) Insulin, Insulinmischungen und Insulinanaloga. Beeinflussen die Insulinwirkung. Hemmen die hepatische Glukose-Freisetzung (Leber) Biguanide. ↓ Insulinresistenz (Leber, Muskeln und Fettgewebe) -Thiazolidinedione und Biguanide. Verlangsamen die Kohlenhydratresorption (Darm) Alphaglucosidase-Inhibitoren. DISKUSSION. Sulfonylharnstoffe und Glinide blockieren den Sulfonylharnstoffrezeptor und die ATP-empfindlichen Kaliumkanäle in den pankreatischen Betazellen. Ihre Hauptnebenwirkungen sind Hypoglykämie und Gewichtszunahme. Insulin entfaltet seine Wirkung über den Insulin-Rezeptor; seine Hauptnebenwirkungen sind Hypoglykämie und Gewichtszunahme. Der Wirkmechanismus der Biguanide (Metformin) ist unbekannt. Ihre wichtigsten Nebenwirkungen sind gastrointestinale Beschwerden und Laktatazidose. Die Thiazolidinedione sind PPAR-gamma-Agonisten. Ihre Hauptnebenwirkungen sind Gewichtszunahme, Ödem und Anämie. Die Alphaglucosidase-Inhibitoren wirken im Darm; ihre wichtigsten Nebenwirkungen sind gastrointestinale Beschwerden. Moller DE. Nature. 2001;414: ; Pickup JC, Williams G, eds. Textbook of Diabetes 2. Malden, MA: Blackwell;2003:45.5.")
25
Alphaglucosidase- Inhibitoren Thiazolidinedione (TZDs)
Medikamente zur Behandlung des Typ 2 Diabetes Medikamenten-klasse Molekulares Ziel Wirkort (e) Nebenwirkungen Sulfonylharnstoffe SU-Rezeptor/K+ ATP-Kanal Pankreatische Betazelle Hypoglykämie Gewichtszunahme Meglitinidanaloga Biguanide Unbekannt Leber Muskeln Gastrointestinale Beschwerden Laktatazidose Alphaglucosidase- Inhibitoren Alphaglucosidase Darm Thiazolidinedione (TZDs) PPAR-gamma(-Agonist) Fettgewebe Ödem Anämie Insulin Insulinrezeptor DISKUSSION Die Insulinsekretagoga (Sulfonylharnstoffe und Glinide) wirken auf die pankreatische Insulinproduktion. Metformin, ein Biguanid, hemmt die Glukosefreisetzung aus der Leber. Sowohl Thiazolidinedione als auch Biguanide verringern die Insulinresistenz. Andere Medikamente verlangsamen die Kohlenhydrat-Resorption aus dem Darm durch Hemmung der Alphaglucosidase. Es gibt Medikamente, die Insulin im Körper ersetzen. Hierzu gehören exogenes Insulin, Insulinmischungen und Insulinanaloga. Moller DE. Nature. 2001;414: ; Pickup JC, Williams G, eds. Textbook of Diabetes 2. Malden, MA: Blackwell;2003:45.5.
 Nebenwirkungen. Sulfonylharnstoffe. SU-Rezeptor/K+ ATP-Kanal. Pankreatische Betazelle. Hypoglykämie. Gewichtszunahme. Meglitinidanaloga. Biguanide. Unbekannt. Leber. Muskeln. Gastrointestinale Beschwerden. Laktatazidose. Alphaglucosidase- Inhibitoren. Alphaglucosidase. Darm. Thiazolidinedione (TZDs) PPAR-gamma(-Agonist) Fettgewebe. Ödem. Anämie. Insulin. Insulinrezeptor. DISKUSSION. Die Insulinsekretagoga (Sulfonylharnstoffe und Glinide) wirken auf die pankreatische Insulinproduktion. Metformin, ein Biguanid, hemmt die Glukosefreisetzung aus der Leber. Sowohl Thiazolidinedione als auch Biguanide verringern die Insulinresistenz. Andere Medikamente verlangsamen die Kohlenhydrat-Resorption aus dem Darm durch Hemmung der Alphaglucosidase. Es gibt Medikamente, die Insulin im Körper ersetzen. Hierzu gehören exogenes Insulin, Insulinmischungen und Insulinanaloga. Moller DE. Nature. 2001;414: ; Pickup JC, Williams G, eds. Textbook of Diabetes 2. Malden, MA: Blackwell;2003:45.5.")
26
Zukünftige Therapien bei Typ 2 Diabetes Gegenwärtige Herausforderungen
Klinische Herausforderungen und zukünftige therapeutische Ansätze Zukünftige Therapien bei Typ 2 Diabetes Unzureichende postprandiale Blutglukoseeinstellung Gewichtszunahme Erhöhtes Hypoglykämierisiko Spezielle Kontraindikationen Zunehmender Verlust der Betazellfunktion Reduzieren – die makrovaskuläre/zerebrovaskuläre Erkr. – das mikrovaskuläre Risiko DISKUSSION Die zukünftigen Therapien für den Typ 2 Diabetes sollten sowohl die makrovaskulären als auch die mikrovaskuläre Risiken im Zusammenhang mit der Erkrankung verringern. Verbessern – die Insulinsekretion und -resistenz – das Sicherheitsprofil – niedriges Hypoglykämierisiko – keine Gewichtszunahme – keine anderen klinisch relevanten Nebenw. Wirken auf die Hyperglykämie – Nüchtern- und postprandiale Wirkungen – Anhaltende Stoffwechseleinstellung im Zeitverlauf Gegenwärtige Herausforderungen

27
Zukünftige Medikamente zur Behandlung des Typ 2 Diabetes
Im Folgenden werden die Merkmale einiger Medikamente diskutiert, die gegenwärtig zur Behandlung des Typ 2 Diabetes entwickelt werden.

28
Die Inkretinhormone sind ein weiterer wesentlicher Bestandteil der normalen Blutglukoseregulation
Als Reaktion auf die Nahrungsaufnahme setzt der Darm Peptidhormone in den Blutkreislauf frei, die auf das Pankreas wirken Diese Hormone werden als Inkretine bezeichnet GLP-1 GIP Inkretinhormone tragen zur Blutglukoseregulation bei durch: Verstärkung der glukoseabhängigen Insulinsekretion Unterdrückung der Glukagonsekretion Regulation der Magenentleerung Regulation der Nahrungsaufnahme DISKUSSION Das Pankreas steuert die Freisetzung der Hormone Insulin und Glukagon, die die Blutglukosespiegel regulieren und zur Blutglukoseregulation beitragen. Im Inselorgan des Pankreas wird Insulin von den Betazellen und Glukagon von den Alphazellen gebildet. Meier JJ, et al. Diabetes Metab Res Rev. 2005; 21(2): ; Holst JJ, et al. Am J Physiol Endocrinol Metab. 2004;287(2):E199-2.
: ; Holst JJ, et al. Am J Physiol Endocrinol Metab. 2004;287(2):E")
29
Probanden ohne Diabetes Patienten mit Diabetes
Inkretinwirkung: Intestinale Faktoren beeinflussen die Insulinsekretion Orale Glukose Intravenöse Glukose 120 Probanden ohne Diabetes 120 Patienten mit Diabetes DISKUSSION Die Plasmainsulinreaktion auf intravenöse Glukose war weniger als halb so groß wie nach oraler Glukoseaufnahme; dies spricht dafür, dass außer der Blutglukose Ernährungsfaktoren (Inkretine) die Insulinsekretion beeinflussen. Inkretine fördern die glukoseabhängige Insulinsekretion. HINTERGRUND Ein Inkretin ist eine Substanz, die vom Darm als Reaktion auf Nahrungsaufnahme in den Blutkreislauf freigesetzt wird. Die Studienstichprobe bestand aus 30 normalgewichtigen Personen (n = 12 mit Typ 2 Diabetes, n = 18 ohne Typ 2 Diabetes). 80 80 Plasmainsulin (U/ml) Plasmainsulin (U/ml) 40 40 -1 1 2 3 4 -1 1 2 3 4 Zeit (Stunden) Zeit (Stunden) Mean (SEM) Modifiziert nach Perley MJ, et al. J Clin Invest. 1967;46:
 die Insulinsekretion beeinflussen. Inkretine fördern die glukoseabhängige Insulinsekretion. HINTERGRUND. Ein Inkretin ist eine Substanz, die vom Darm als Reaktion auf Nahrungsaufnahme in den Blutkreislauf freigesetzt wird. Die Studienstichprobe bestand aus 30 normalgewichtigen Personen (n = 12 mit Typ 2 Diabetes, n = 18 ohne Typ 2 Diabetes) Plasmainsulin (U/ml) Plasmainsulin (U/ml) Zeit (Stunden) Zeit (Stunden) Mean (SEM) Modifiziert nach Perley MJ, et al. J Clin Invest. 1967;46:")
30
Die GLP-1-Wirkungen sind beim Typ 2 Diabetes glukoseabhängig
Placebo GLP-1 PBO PBO PBO GLP-1 GLP-1 GLP-1 270 300 20 DISKUSSION Eine Dauerinfusion von GLP-1 führte während eines 4-stündigen Zeitraums zu einer signifikanten Blutglukosesenkung, verglichen mit Placebo. im Vergleich zu Placebo verstärkte GLP-1 anfangs die Insulinsekretion; wenn die Blutglukose normale Konzentrationen erreichte, kam die Insulinsekretion jedoch trotz der fortgeführten GLP-1-Dauerinfusion wieder auf Baseline-Niveau – dies ist ein Beweis für eine glukoseabhängige Insulinsekretion. GLP-1 senkt die Glukagonkonzentrationen bei Vorliegen einer Hyperglykämie. Die Glukagonspiegel steigen jedoch trotz fortgesetzter GLP-1-Infusion wieder auf die Ausgangswerte an, wenn sich die Blutglukose normalisiert; dies zeigt, dass GLP-1 Glukagon bei Euglykämie oder Hypoglykämie nicht senkt. Die Glukoseabhängigkeit wird durch einen Rückgang von Plasmainsulin und -glukagon auf Konzentrationen wie vor Behandlungsbeginn nachgewiesen, wenn die Blutglukose den Normalbereich erreicht. HINTERGRUND In diese Studie waren Probanden mit Typ 2 Diabetes (n = 10) eingeschlossen – alle erhielten Diät und einen Sulfonylharnstoff (SFU). Zudem wurden einige Probanden mit Metformin oder Acarbose behandelt. Alle Antidiabetika wurden bei Studienbeginn abgesetzt. Intravenöses GLP-1(7-36)amid wurde 4 Stunden lang mit 1,2 pmol/kg/min infundiert. * 180 200 Glukose (mg/dl) Insulin (pmol/l) * * Glukagon (pmol/l) 10 * * * * * 90 100 * * * * * -30 60 120 180 240 -30 60 120 180 240 -30 60 120 180 240 Zeit (min) Zeit (min) Zeit (min) n= 10; Mittelwert±SEM; *p < 0,05. Nauck MA, et al. Diabetologia. 1993;36: Nachdruck mit Erlaubnis des Springer-Verlags © 1993.
 eingeschlossen – alle erhielten Diät und einen Sulfonylharnstoff (SFU). Zudem wurden einige Probanden mit Metformin oder Acarbose behandelt. Alle Antidiabetika wurden bei Studienbeginn abgesetzt. Intravenöses GLP-1(7-36)amid wurde 4 Stunden lang mit 1,2 pmol/kg/min infundiert. * Glukose (mg/dl) Insulin (pmol/l) * * Glukagon (pmol/l) 10. * * * * * * * * * * Zeit (min) Zeit (min) Zeit (min) n= 10; Mittelwert±SEM; *p < 0,05. Nauck MA, et al. Diabetologia. 1993;36: Nachdruck mit Erlaubnis des Springer-Verlags ©")
31
Das therapeutische Potenzial von GLP-1 wird durch seine schnelle Inaktivierung begrenzt
Schnelle Inaktivierung (DPP-IV), Kurze Eliminationshalbwertszeit (ca. 1–2 min) GLP-1 muss kontinuierlich verabreicht werden (Infusion) DISKUSSION Trotz seiner günstigen biologischen Wirkungen ist das therapeutische Potenzial von GLP-1 vor allem durch seinen schnellen Abbau durch das ubiquitäre Enzym Dipeptidyl-Peptidase-IV (DPP-IV) begrenzt. Die schnelle Inaktivierung von GLP-1 trägt neben seiner schnellen renalen Clearance zu der kurzen Halbwertszeit von weniger als 2 Minuten bei. Daher ist für die Aufrechterhaltung von Plasmakonzentrationen von GLP-1, die lange genug sind, um eine therapeutische Wirkung hervorzurufen, eine Dauerverabreichung erforderlich. Wie dies bei anderen Peptiden typisch ist, muss auch GLP-1 injiziert werden. Unpraktisch für die Behandlung einer chronischen Krankheit wie dem Typ 2 Diabetes Drucker DJ, et al. Diabetes Care. 2003;26:
, Kurze Eliminationshalbwertszeit (ca. 1–2 min) GLP-1 muss kontinuierlich verabreicht werden (Infusion) DISKUSSION. Trotz seiner günstigen biologischen Wirkungen ist das therapeutische Potenzial von GLP-1 vor allem durch seinen schnellen Abbau durch das ubiquitäre Enzym Dipeptidyl-Peptidase-IV (DPP-IV) begrenzt. Die schnelle Inaktivierung von GLP-1 trägt neben seiner schnellen renalen Clearance zu der kurzen Halbwertszeit von weniger als 2 Minuten bei. Daher ist für die Aufrechterhaltung von Plasmakonzentrationen von GLP-1, die lange genug sind, um eine therapeutische Wirkung hervorzurufen, eine Dauerverabreichung erforderlich. Wie dies bei anderen Peptiden typisch ist, muss auch GLP-1 injiziert werden. Unpraktisch für die Behandlung einer chronischen Krankheit wie dem. Typ 2 Diabetes. Drucker DJ, et al. Diabetes Care. 2003;26:")
32
Gegenwärtige Ansätze auf GLP-1-Basis zur Verbesserung der Blutglukose-Einstellung
Medikamente, die die Wirkungen von GLP-1 nachahmen (Inkretin-Mimetika) DPP-IV-resistente GLP-1-Derivate Beispiele: GLP-1-Analoga, Albumin-gebundenes GLP-1 Neue Peptide, die die blutzuckerregulierenden Wirkungen von GLP-1 nachahmen Exenatide Medikamente, die die Wirkung von endogenem GLP-1 verlängern DPP-IV-Inhibitoren Drucker DJ, et al. Diabetes Care. 2003;26: ; Baggio LL, et al. Diabetes. 2004; 53:
 DPP-IV-resistente GLP-1-Derivate. Beispiele: GLP-1-Analoga, Albumin-gebundenes GLP-1. Neue Peptide, die die blutzuckerregulierenden Wirkungen von GLP-1 nachahmen. Exenatide. Medikamente, die die Wirkung von endogenem GLP-1 verlängern. DPP-IV-Inhibitoren. Drucker DJ, et al. Diabetes Care. 2003;26: ; Baggio LL, et al. Diabetes. 2004; 53:")
33
Entwicklung von Exenatide: Ein Inkretin-Mimetikum
Exenatide (Exendin-4) Synthetische Version des Speichelproteins (in der Gila-Krustenechse nachgewiesen) Ca. 50%ige Übereinstimmung mit dem humanen GLP-1 Bindet an bekannte humane GLP-1-Rezeptoren auf Betazellen in vitro Resistent gegen Inaktivierung durch DPP-IV DISKUSSION Exenatide ist ein Inkretin-Mimetikum, das aus einem Naturprodukt entwickelt wurde, welches in der Gila-Krustenechse nachgewiesen wurde. Die Aminosäure in Position 2, dem Ort der DPP-IV-Inaktivierung auf dem GLP-1-Molekül, ist bei Exenatide eine andere; dadurch wird Exenatide gegenüber dem enzymatischen Abbau durch DPP-IV resistent. HINTERGRUND Nach subkutaner Verabreichung an Patienten mit Typ 2 Diabetes erreicht Exenatide innerhalb von 2-3 Stunden maximale Plasma-Konzentrationen. Die terminale Halbwertzeit von Exenatide beträgt ca. 2-6 Stunden. Exenatide und endogenes GLP-1 besitzen einige gemeinsame Blutzucker-regulierende Wirkungen. H G E G T F T S D L S K Q M E E E A V R L F I E W L K N G G P S S G A P P P S – NH2 H A E G T F T S D V S S Y L E G Q A A K E F I A W L V K G R – NH2 Exenatide GLP-1 Human Ort der Inaktivierung durch DPP-IV Modifiziert nach Nielsen LL, et al. Regulatory Peptides. 2004;117:77-88.; Fineman MS, et al. Diabetes Care. 2003;26: Nachdruck aus Pharmacology of Exenatidee (synthetic exendin-4): a potential therapeutic for improved glycemic control of type 2 diabetes, 77-88, Copyright 2004, mit Erlaubnis von Elsevier.
 Synthetische Version des Speichelproteins (in der Gila-Krustenechse nachgewiesen) Ca. 50%ige Übereinstimmung mit dem humanen GLP-1. Bindet an bekannte humane GLP-1-Rezeptoren auf Betazellen in vitro. Resistent gegen Inaktivierung durch DPP-IV. DISKUSSION. Exenatide ist ein Inkretin-Mimetikum, das aus einem Naturprodukt entwickelt wurde, welches in der Gila-Krustenechse nachgewiesen wurde. Die Aminosäure in Position 2, dem Ort der DPP-IV-Inaktivierung auf dem GLP-1-Molekül, ist bei Exenatide eine andere; dadurch wird Exenatide gegenüber dem enzymatischen Abbau durch DPP-IV resistent. HINTERGRUND. Nach subkutaner Verabreichung an Patienten mit Typ 2 Diabetes erreicht Exenatide innerhalb von 2-3 Stunden maximale Plasma-Konzentrationen. Die terminale Halbwertzeit von Exenatide beträgt ca. 2-6 Stunden. Exenatide und endogenes GLP-1 besitzen einige gemeinsame Blutzucker-regulierende Wirkungen. H G E G T F T S D L S K Q M E E E A V R L F I E W L K N G G P S S G A P P P S – NH2. H A E G T F T S D V S S Y L E G Q A A K E F I A W L V K G R – NH2. Exenatide. GLP-1. Human. Ort der Inaktivierung durch DPP-IV. Modifiziert nach Nielsen LL, et al. Regulatory Peptides. 2004;117:77-88.; Fineman MS, et al. Diabetes Care. 2003;26: Nachdruck aus Pharmacology of Exenatidee (synthetic exendin-4): a potential therapeutic for improved glycemic control of type 2 diabetes, 77-88, Copyright 2004, mit Erlaubnis von Elsevier.")
34
Merkmale von GLP-1 versus Exenatide
Verstärkt die Glukose-abhängige Insulin-Sekretion Hemmt übermäßig hohe Glukagon-Sekretion Verlangsamt die Magenentleerung Verringert die Nahrungsaufnahme und hemmt die Gewichtszunahme Stimuliert die Neogenese und die Proliferation von Insulin-produzierenden Zellen bei Tieren Resistent gegen Abbau durch DPP-IV Plasma-Halbwertzeit nach s.c.-Injektion <2 min min DISKUSSION GLP-1 und Exenatide haben zahlreiche klinische relevante gemeinsame Merkmale, Exenatide stellt jedoch eine therapeutisch besser anwendbare Möglichkeit dar, da es gegen den Abbau durch das Enzym Dipeptidyl-Peptidase-IV (DPP-IV) resistent ist und daher eine längere Halbwertzeit besitzt. Drucker DJ. Diabetes Care. 2003;26: ; Fineman MS, et al. Diabetes Care. 2003;26: ; Zander M, et al. Lancet 2002;359:
 resistent ist und daher eine längere Halbwertzeit besitzt. Drucker DJ. Diabetes Care. 2003;26: ; Fineman MS, et al. Diabetes Care. 2003;26: ; Zander M, et al. Lancet 2002;359:")
35
Große klinische Phase-3-Studien
Exenatide versus Placebo Hinzugefügt zu maximal wirksamen Dosen von Metformin (MET) und/oder Sulfonylharnstoff (SFU) bei Patienten mit Typ-2-Diabetes MET (n=336) SFU (n=377) MET + SFU (n=733) DISKUSSION Drei 30-wöchige, doppelblinde pivotale Phase-3-Studien wurden in den Vereinigten Staaten durchgeführt. In diesen Studien wurde Exenatidee den bereits verabreichten maximal wirksamen Dosen einer Behandlung mit Metformin (MET), eines Sulfonylharnstoffs (SFU) oder MET + SFU Bei Patienten mit Typ 2 Diabetes mit HbA1C-Werten über 7% hinzugefügt. Exenatide + MET Studie: mindestens 1500 mg MET pro Tag, 336 Patienten Exenatide + SFU Studie: mindestens die maximal wirksame Dosis von SFU allein, 377 Patienten Exenatide + MET + SFU Studie: MET + SFU, 733 Patienten HINTERGRUND Die drei großen klinischen Phase-3-Studien wurden auch als die drei AMIGOs bezeichnet: AC2993: Diabetes Management for Improving Glucose Outcomes [Diabetesbehandlung zur Verbesserung der Blutglukosewerte] DeFronzo RA, et al. Diabetes Care. 2005;28: ; Buse JB, et al. Diabetes Care. 2004;27: ; Kendall DM, et al. Diabetes Care. 2005;28:
 und/oder Sulfonylharnstoff (SFU) bei Patienten mit Typ-2-Diabetes. MET (n=336) SFU (n=377) MET + SFU (n=733) DISKUSSION. Drei 30-wöchige, doppelblinde pivotale Phase-3-Studien wurden in den Vereinigten Staaten durchgeführt. In diesen Studien wurde Exenatidee den bereits verabreichten maximal wirksamen Dosen einer Behandlung mit Metformin (MET), eines Sulfonylharnstoffs (SFU) oder MET + SFU Bei Patienten mit Typ 2 Diabetes mit HbA1C-Werten über 7% hinzugefügt. Exenatide + MET Studie: mindestens 1500 mg MET pro Tag, 336 Patienten. Exenatide + SFU Studie: mindestens die maximal wirksame Dosis von SFU allein, 377 Patienten. Exenatide + MET + SFU Studie: MET + SFU, 733 Patienten. HINTERGRUND. Die drei großen klinischen Phase-3-Studien wurden auch als die drei AMIGOs bezeichnet: AC2993: Diabetes Management for Improving Glucose Outcomes [Diabetesbehandlung zur Verbesserung der Blutglukosewerte] DeFronzo RA, et al. Diabetes Care. 2005;28: ; Buse JB, et al. Diabetes Care. 2004;27: ; Kendall DM, et al. Diabetes Care. 2005;28:")
36
Studiendesign der großen klinischen Phase-3-Studien
Randomisierte, doppelblinde, placebokontrollierte, multi-zentrische Studien an Patienten mit Typ 2 Diabetes Keine Auswaschphase Verabreichung von Exenatide oder Placebo subkutan vor dem Frühstück und dem Abendessen DISKUSSION Die drei AMIGOs waren randomisierte, doppelblinde, placebokontrollierte, multizentrische Studien. Die Patienten wurden für einen von vier Behandlungsarmen nach einer 4-wöchigen Einleitungsphase mit Injektion von 0,02 ml Placebo randomisiert: 5 µg Exenatide für die Dauer der Studie 5 µg von Exenatide für die nächsten 4 Wochen und anschließend 10 µg Exenatide für die restliche Dauer der 30-wöchigen Studie Eine Injektion vom 0,02 ml Placebo für die Dauer der Studie Eine Injektion vom 0,02 ml Placebo für die nächsten 4 Wochen und anschließend eine Injektion von 0,04 ml Placebo für die restliche Dauer der 30-wöchigen Studie HINTERGRUND Die Patienten erhielten die Instruktion, die Einnahme ihrer oralen Diabetes-Medikamente fortzusetzen; dabei wurde Exenatide als Zusatztherapie zu den bestehenden Medikamenten-Schemata hinzugefügt. Exenatide wurde von den Patienten 2xtgl subkutan 15 Minuten vor dem Frühstück und dem Abendessen selbst verabreicht. Beide Exenatide-Arme begannen mit 5 µg Exenatide für 4 Wochen, um die Übelkeit auf ein Minimum zu reduzieren; außerdem wurden die Placebogruppen für die Analysen zusammengefasst. Die Teilnehmer konnten die Studie unter folgenden Bedingungen vorzeitig beenden: Abweichung des HbA1c von bis zu 1,5% vom Ausgangswert oder HbA1c >11,5% in Woche 18 oder 24 oder Nüchtern-Blutglukose >240 mg/dl (Labortest) oder Nüchtern-Blutglukose >260 mg/dl (Fingerbeere) über mindestens 2 Wochen Exenatide 5 µg (0,02 ml) 2xtgl Exenatide 5 µg (0,02 ml) 2xtgl Placebo- Einleitung 0,02 ml 2xtgl Exenatide 10 µg (0,04 ml) 2xtgl Screening Placebo0,02 ml 2xtgl Placebo 5 µg (0,02 ml) oder 10 µg (0,04 ml) 2xtgl -4 4 12 24 30 Zeit (Wochen) DeFronzo RA, et al. Diabetes Care. 2005;28: ; Buse JB, et al. Diabetes Care. 2004;27: ; Kendall DM, et al. Diabetes Care. 2005;28:
 oder. Nüchtern-Blutglukose >260 mg/dl (Fingerbeere) über mindestens 2 Wochen. Exenatide 5 µg (0,02 ml) 2xtgl. Exenatide 5 µg (0,02 ml) 2xtgl. Placebo- Einleitung 0,02 ml 2xtgl. Exenatide 10 µg (0,04 ml) 2xtgl. Screening. Placebo0,02 ml 2xtgl. Placebo 5 µg (0,02 ml) oder 10 µg (0,04 ml) 2xtgl Zeit (Wochen) DeFronzo RA, et al. Diabetes Care. 2005;28: ; Buse JB, et al. Diabetes Care. 2004;27: ; Kendall DM, et al. Diabetes Care. 2005;28:")
37
Demographische Merkmale der Patienten in den großen klinischen Phase-3-Studien
Race (%) Age (y) Gender, male/female (%) Rasse (%) (weiß/schwarz/hispanisch/andere) Alter (J) Geschlecht, m / w (%) BMI (kg/m2) Diabetesdauer (J) HbA1C (%) Gewicht (kg) ITT 69 / 12 / 16 / 3 55 10 58 / 42 n=483 8 6 8,5 1,1 34 5 99 24 Placebo 2xtgl 5 µg Exenatidee 2xtgl 10 µg Exenatidee 2xtgl n=480 n=483 58 / 42 59 / 41 59 / 41 DISKUSSION Bei allen drei Studien und in allen drei Behandlungsarmen waren die demographischen Merkmale der Patienten ähnlich. Es bestand eine ähnliche Aufteilung der Patienten hinsichtlich Männern und Frauen sowie der rassischen Zugehörigkeit. Der typische Studienpatient war ein Mittfünfziger mit einem Gewicht von 100 kg und einem Body-Mass-Index (BMI) von 33 bis 34 kg/m2, einem Ausgangs-HbA1c von ungefähr 8,5% und einer Diabetesdauer von fast 8 Jahren. HINTERGRUND Einschlusskriterien für die AMIGO-Studien (30-wöchige, doppelblinde, Phase-3-Studien): Patienten mit Typ 2 Diabetes, die mit Metformin (MET), einem Sulphonylharnstoff (SFU), oder MET+SFU behandelt wurden Alter von 16 bis 75 Jahren, HbA1c 7,1% bis 11,0%, Nüchtern-Blutzucker <240 mg/dl, BMI 27 bis 45 kg/m2 Stabiles Gewicht (±10%) über 3 Monate vor dem Screening Randomisierte Patienten waren definiert als alle Patienten, denen eine Randomisierungsnummer zugeordnet wurden: Placebo (n=483), 5 µg 2xtgl (n=480) oder 10 µg 2xtgl (n=484) Intent-to-treat- (ITT-) Patienten waren definiert als alle randomisierten Patienten, die mindestens eine Injektion der randomisierte Medikation erhielten. 55 10 55 11 69 / 12 / 15 / 4 68 / 12 / 16 / 4 97 20 98 20 33 6 34 34 6 8,4 1,1 8,5 1.1 1,1 8 6 7 6 ITT combined; Mean (SD) unless otherwise specified DeFronzo RA, et al. Diabetes Care. 2005;28: ; Buse JB, et al. Diabetes Care. 2004;27: ; Kendall DM, et al. Diabetes Care. 2005;28:
 Age (y) Gender, male/female (%) Rasse (%) (weiß/schwarz/hispanisch/andere) Alter (J) Geschlecht, m / w (%) BMI (kg/m2) Diabetesdauer (J) HbA1C (%) Gewicht (kg) ITT. 69 / 12 / 16 / / 42. n= ,5. 1, Placebo 2xtgl. 5 µg Exenatidee 2xtgl. 10 µg Exenatidee 2xtgl. n=480. n= / / / 41. DISKUSSION. Bei allen drei Studien und in allen drei Behandlungsarmen waren die demographischen Merkmale der Patienten ähnlich. Es bestand eine ähnliche Aufteilung der Patienten hinsichtlich Männern und Frauen sowie der rassischen Zugehörigkeit. Der typische Studienpatient war ein Mittfünfziger mit einem Gewicht von 100 kg und einem Body-Mass-Index (BMI) von 33 bis 34 kg/m2, einem Ausgangs-HbA1c von ungefähr 8,5% und einer Diabetesdauer von fast 8 Jahren. HINTERGRUND. Einschlusskriterien für die AMIGO-Studien (30-wöchige, doppelblinde, Phase-3-Studien): Patienten mit Typ 2 Diabetes, die mit Metformin (MET), einem Sulphonylharnstoff (SFU), oder MET+SFU behandelt wurden. Alter von 16 bis 75 Jahren, HbA1c 7,1% bis 11,0%, Nüchtern-Blutzucker <240 mg/dl, BMI 27 bis 45 kg/m2. Stabiles Gewicht (±10%) über 3 Monate vor dem Screening. Randomisierte Patienten waren definiert als alle Patienten, denen eine Randomisierungsnummer zugeordnet wurden: Placebo (n=483), 5 µg 2xtgl (n=480) oder 10 µg 2xtgl (n=484) Intent-to-treat- (ITT-) Patienten waren definiert als alle randomisierten Patienten, die mindestens eine Injektion der randomisierte Medikation erhielten / 12 / 15 / / 12 / 16 / ,4. 1,1. 8, , ITT combined; Mean (SD) unless otherwise specified DeFronzo RA, et al. Diabetes Care. 2005;28: ; Buse JB, et al. Diabetes Care. 2004;27: ; Kendall DM, et al. Diabetes Care. 2005;28:")
38
Aufteilung der Patienten in den großen klinischen Phase-3-Studien
Placebo 2xtgl 5 µg Exenatidee 2xtgl 10 µg Exenatidee 2xtgl ITT n=483 n=480 n=483 Abgeschlossen 351 (73%) 391 (81%) 383 (79%) DISKUSSION In jedem Behandlungsarm schloss eine ähnliche Zahl von Patienten die Phase-3-Studien ab; die Gesamtabschlussrate betrug 78%. In der Placebo-Behandlungsgruppe war die Abbruchrate am höchsten – hauptsächlich wegen des Verlust der Stoffwechselregulation (da ihr Diabetes mit ihrem/n Ausgangsmedikament(en) unzureichend eingestellt war) und wegen Widerrufs der Einwilligung. In den Exenatide-Behandlungsgruppen brachen mehr Patienten die Studie aufgrund von Nebenwirkungen ab, die zumeist gastrointestinaler Art waren, als in den Placebogruppen. HINTERGRUND Drei 30-wöchige, placebokontrollierte, doppelblinde Phase-3-Studien; Patienten mit Typ 2 Diabetes, die für Placebo oder 5 µg oder 10 µg Exenatide 2xtgl mit MET und/oder SFU, n=1446, randomisiert waren Als Verlust der Stoffwechselkontrolle galt das Zutreffen mindestens eines der folgenden Kriterien: Anstieg des HbA1 c gegenüber dem Ausgangswert um 1,5% oder HbA1c 11,5% zu bestimmten Zeitpunkten Nüchtern-Blutglukose (NBG) >240 mg/dl bei zwei aufeinander folgenden Klinikvisiten Selbst gemessener NBG >260 mg/dl über mindestens 2 Wochen (falls auf eine Krankheit oder eine Medikation zurückzuführen) Widerruf der Einwilligung: allen Probanden war das Recht vorbehalten, ohne jegliche Strafe oder jeglichen Verlust von Vorteilen aus der Studie auszuscheiden. Abbruch 132 (27%) 89 (19%) 101 (21%) Nebenwirkungen 3,3% 5,6% 8,9% Verlust der Stoffwechselkontrolle 7,2% 3,1% 1,9% Aus der Beobachtung gefallen 3,3% 2,9% 3,1% Widerruf der Einwilligung 9,1% 5,2% 5,2% ITT zusammengefasst; Mittelwert (SD) sofern nicht anders angegeben DeFronzo RA, et al. Diabetes Care. 2005;28: ; Buse JB, et al. Diabetes Care. 2004;27: ; Kendall DM, et al. Diabetes Care. 2005;28:
 391 (81%) 383 (79%) DISKUSSION. In jedem Behandlungsarm schloss eine ähnliche Zahl von Patienten die Phase-3-Studien ab; die Gesamtabschlussrate betrug 78%. In der Placebo-Behandlungsgruppe war die Abbruchrate am höchsten – hauptsächlich wegen des Verlust der Stoffwechselregulation (da ihr Diabetes mit ihrem/n Ausgangsmedikament(en) unzureichend eingestellt war) und wegen Widerrufs der Einwilligung. In den Exenatide-Behandlungsgruppen brachen mehr Patienten die Studie aufgrund von Nebenwirkungen ab, die zumeist gastrointestinaler Art waren, als in den Placebogruppen. HINTERGRUND. Drei 30-wöchige, placebokontrollierte, doppelblinde Phase-3-Studien; Patienten mit Typ 2 Diabetes, die für Placebo oder 5 µg oder 10 µg Exenatide 2xtgl mit MET und/oder SFU, n=1446, randomisiert waren. Als Verlust der Stoffwechselkontrolle galt das Zutreffen mindestens eines der folgenden Kriterien: Anstieg des HbA1 c gegenüber dem Ausgangswert um 1,5% oder HbA1c 11,5% zu bestimmten Zeitpunkten. Nüchtern-Blutglukose (NBG) >240 mg/dl bei zwei aufeinander folgenden Klinikvisiten. Selbst gemessener NBG >260 mg/dl über mindestens 2 Wochen (falls auf eine Krankheit oder eine Medikation zurückzuführen) Widerruf der Einwilligung: allen Probanden war das Recht vorbehalten, ohne jegliche Strafe oder jeglichen Verlust von Vorteilen aus der Studie auszuscheiden. Abbruch. 132 (27%) 89 (19%) 101 (21%) Nebenwirkungen. 3,3% 5,6% 8,9% Verlust der Stoffwechselkontrolle. 7,2% 3,1% 1,9% Aus der. Beobachtung gefallen. 3,3% 2,9% 3,1% Widerruf der Einwilligung. 9,1% 5,2% 5,2% ITT zusammengefasst; Mittelwert (SD) sofern nicht anders angegeben DeFronzo RA, et al. Diabetes Care. 2005;28: ; Buse JB, et al. Diabetes Care. 2004;27: ; Kendall DM, et al. Diabetes Care. 2005;28:")
39
Exenatide senkte HbA1C und Gewicht: Große (zusammengefasste) klin
Exenatide senkte HbA1C und Gewicht: Große (zusammengefasste) klin. Phase-3-Studien Placebo 2xtgl 5 µg Exenatide 2xtgl 10 µg Exenatide 2xtgl -0,5 0,5 0,1 DISKUSSION Die zusammengefassten Ergebnisse der drei klinischen Phase-3-Studien zeigten, dass Exenatidee die Blutzucker-Einstellung verbesserte und das Körpergewicht senkte. Der mittlere HbA1c wurde (im Vergleich zum Ausgangswert): um +0,1% in der Placebogruppe erhöht um -0,6% in der 5-µg Behandlungsgruppe und um -0,9% in der 10-µg Behandlungsgruppe gesenkt Das mittlere Körpergewicht wurde (im Vergleich zum Ausgangswert): um -0,6 kg in der Placebogruppe gesenkt um -1,4 kg in der 5-µg Behandlungsgruppe und um -1,9 in der 10-µg Behandlungsgruppe gesenkt HINTERGRUND HbA1c-Ausgangswert: Placebo, 8,5%; 5 µg 2xtgl, 8,4%; 10 µg 2xtgl, 8,5%. Ausgangsgewicht: Placebo, 99 kg; 5 µg 2xtgl, 96 kg; 10 µg 2xtgl, 98 kg. Zusammengefasste AMIGO-Studien: drei 30-wöchige, doppelblinde Phase-3-Studien Patienten mit Typ 2 Diabetes, die mit Metformin (MET), einem Sulfonylharnstoff (SFU) oder MET + SFU behandelt wurden Randomisiert für Placebo (n=483), 5 µg 2xtgl (n=480) oder 10 µg 2xtgl (n=483) plus bereits eingeleitete Diabetes-Behandlung. Die Gewichtsänderung war ein sekundärer Endpunkt. -0,5 Weight (kg) HbA1C (%) -0,5 -1 -0,7 -0,6 * -1 -0,9 * -1,5 -1,4 * -1,5 -2 -1,9 * ITT-Daten nach 30 Wochen; N = 1446; Mittelwert (SE); *P<0,005; das Gewicht war ein sekundärer Endpunkt Interne Daten, Amylin Pharmaceuticals, Inc.
 klin. Phase-3-Studien. Placebo 2xtgl 5 µg Exenatide 2xtgl 10 µg Exenatide 2xtgl. -0,5. 0,5. 0,1. DISKUSSION. Die zusammengefassten Ergebnisse der drei klinischen Phase-3-Studien zeigten, dass Exenatidee die Blutzucker-Einstellung verbesserte und das Körpergewicht senkte. Der mittlere HbA1c wurde (im Vergleich zum Ausgangswert): um +0,1% in der Placebogruppe erhöht. um -0,6% in der 5-µg Behandlungsgruppe und um -0,9% in der 10-µg Behandlungsgruppe gesenkt. Das mittlere Körpergewicht wurde (im Vergleich zum Ausgangswert): um -0,6 kg in der Placebogruppe gesenkt. um -1,4 kg in der 5-µg Behandlungsgruppe und um -1,9 in der 10-µg Behandlungsgruppe gesenkt. HINTERGRUND. HbA1c-Ausgangswert: Placebo, 8,5%; 5 µg 2xtgl, 8,4%; 10 µg 2xtgl, 8,5%. Ausgangsgewicht: Placebo, 99 kg; 5 µg 2xtgl, 96 kg; 10 µg 2xtgl, 98 kg. Zusammengefasste AMIGO-Studien: drei 30-wöchige, doppelblinde Phase-3-Studien. Patienten mit Typ 2 Diabetes, die mit Metformin (MET), einem Sulfonylharnstoff (SFU) oder MET + SFU behandelt wurden. Randomisiert für Placebo (n=483), 5 µg 2xtgl (n=480) oder 10 µg 2xtgl (n=483) plus bereits eingeleitete Diabetes-Behandlung. Die Gewichtsänderung war ein sekundärer Endpunkt. -0,5. Weight (kg) HbA1C (%) -0, ,7. -0,6. * ,9. * -1,5. -1,4. * -1, ,9. * ITT-Daten nach 30 Wochen; N = 1446; Mittelwert (SE); *P<0,005; das Gewicht war ein sekundärer Endpunkt Interne Daten, Amylin Pharmaceuticals, Inc.")
40
Häufige Nebenwirkungen in der großen klinischen Phase-3-Studien
Zusammengefasste Ergebnisse der 30-wöchigen Phase-3-Studien zu Exenatide Placebo (n=483) 5 µg Exenatide (n=480) 10 µg Exenatide (n=483) 48% 39% 18% Übelkeit DISKUSSION Die Auswertung der zusammengefassten Ergebnisse aus allen drei klinischen Phase-3-Studien ergab, dass die häufigsten Nebenwirkungen Übelkeit und Hypoglykämie waren. In der Regel war die Übelkeit leicht bis mäßig ausgeprägt und hatte geringe oder mäßige Auswirkungen auf die Alltagsaktivitäten. Die Inzidenz der Übelkeit nahm im Zeitverlauf ab. Auch die Hypoglykämie war zumeist leicht bis mäßig ausgeprägt. Es trat ein schwerwiegendes hypoglykämisches Ereignis auf (im 5 µg-Exenatide- Behandlungsarm). Exenatide erhöhte die Inzidenz der Hypoglykämie nicht, wenn es mit Metformin (MET) kombiniert wurde. Die Inzidenz der leichten bis mäßigen Hypoglykämie stieg in beiden Gruppen an, wenn Exenatide einem Sulfonylharnstoff (SFU) hinzugefügt wurde. HINTERGRUND Drei 30-wöchige, doppelblinde, Phase-3-Studien wurden in den Vereinigten Staaten abgeschlossen: Patienten mit Typ 2 Diabetes randomisiert für Placebo, 5 µg Exenatide 2xtgl, oder 10 µg Exenatide 2xtgl mit MET, ITT n=336 Patienten mit Typ 2 Diabetes randomisiert für Placebo, 5 µg Exenatide 2xtgl, oder 10 µg Exenatide 2xtgl mit SFU, ITT n=377 Patienten mit Typ 2 Diabetes randomisiert für Placebo, 5 µg Exenatide 2xtgl, oder 10 µg Exenatide 2xtgl mit MET und SFU, ITT n=733 HYPOGLYKÄMIE-DEFINITIONEN Leichte bis mäßige Hypoglykämie: von den Probanden angegebene, mit Hypoglykämie vereinbare Symptome, die mit einem Blutzucker-Wert (<3,33 mmol/l) dokumentiert worden wären. Schwere Hypoglykämie: Patient benötigte die Hilfe einer anderen Person zur Behandlung der Hypoglykämie; als Behandlung waren orale Kohlenhydrate, intravenöse Glukose oder intramuskuläres Glukagon möglich. 25% 15% 8% Hypoglykämie 15% 11% 6% Diarrhö 13% 4% Erbrechen 7% 10% 6% Kopfschmerzen 10% 9% 4% Zitterigkeit Interne 30-Wochen-Daten, Amylin Pharmaceuticals, Inc.
 5 µg Exenatide (n=480) 10 µg Exenatide (n=483) 48% 39% 18% Übelkeit. DISKUSSION. Die Auswertung der zusammengefassten Ergebnisse aus allen drei klinischen Phase-3-Studien ergab, dass die häufigsten Nebenwirkungen Übelkeit und Hypoglykämie waren. In der Regel war die Übelkeit leicht bis mäßig ausgeprägt und hatte geringe oder mäßige Auswirkungen auf die Alltagsaktivitäten. Die Inzidenz der Übelkeit nahm im Zeitverlauf ab. Auch die Hypoglykämie war zumeist leicht bis mäßig ausgeprägt. Es trat ein schwerwiegendes hypoglykämisches Ereignis auf (im 5 µg-Exenatide- Behandlungsarm). Exenatide erhöhte die Inzidenz der Hypoglykämie nicht, wenn es mit Metformin (MET) kombiniert wurde. Die Inzidenz der leichten bis mäßigen Hypoglykämie stieg in beiden Gruppen an, wenn Exenatide einem Sulfonylharnstoff (SFU) hinzugefügt wurde. HINTERGRUND. Drei 30-wöchige, doppelblinde, Phase-3-Studien wurden in den Vereinigten Staaten abgeschlossen: Patienten mit Typ 2 Diabetes randomisiert für Placebo, 5 µg Exenatide 2xtgl, oder 10 µg Exenatide 2xtgl mit MET, ITT n=336. Patienten mit Typ 2 Diabetes randomisiert für Placebo, 5 µg Exenatide 2xtgl, oder 10 µg Exenatide 2xtgl mit SFU, ITT n=377. Patienten mit Typ 2 Diabetes randomisiert für Placebo, 5 µg Exenatide 2xtgl, oder 10 µg Exenatide 2xtgl mit MET und SFU, ITT n=733. HYPOGLYKÄMIE-DEFINITIONEN. Leichte bis mäßige Hypoglykämie: von den Probanden angegebene, mit Hypoglykämie vereinbare Symptome, die mit einem Blutzucker-Wert (<3,33 mmol/l) dokumentiert worden wären. Schwere Hypoglykämie: Patient benötigte die Hilfe einer anderen Person zur Behandlung der Hypoglykämie; als Behandlung waren orale Kohlenhydrate, intravenöse Glukose oder intramuskuläres Glukagon möglich. 25% 15% 8% Hypoglykämie. 15% 11% 6% Diarrhö. 13% 4% Erbrechen. 7% 10% 6% Kopfschmerzen. 10% 9% 4% Zitterigkeit. Interne 30-Wochen-Daten, Amylin Pharmaceuticals, Inc.")
41
Zusammenfassung Die gegenwärtig verfügbaren Medikamente zur Behandlung des Typ 2 Diabetes weisen erhebliche Beschränkungen auf GLP-1 besitzt einige glukoregulatorische Wirkungen, die für die Behandlung des Typ 2 Diabetes günstig sein könnten Unpraktisch bezüglich der Dauerverabreichung Wirkstoffe, die die Wirkungen von GLP-1 nachahmen oder verstärken, sind von hohem klinischem Interesse

42
Inkretinverstärkung durch DPP-4- Hemmung

43
Veränderte ß-Zell-Funktion bei Typ-2-Diabetes
Verschlechterte oszillatorische Insulinfreigabe Erhöhte Proinsulinspiegel Verlust der 1st-phase Insulinantwort Abnormale 2nd-phase Insulinantwort Progressiver Verlust an ß-Zell-Masse Adapted from Buchanan TA Clin Ther 2003;25(suppl B):B32–B46; Polonsky KS et al N Engl J Med 1988;318:1231–1239; Quddusi S et al Diabetes Care 2003;26:791–798; Porte D Jr, Kahn SE Diabetes 2001;50(suppl 1):S160–S163.
:B32–B46; Polonsky KS et al N Engl J Med 1988;318:1231–1239; Quddusi S et al Diabetes Care 2003;26:791–798; Porte D Jr, Kahn SE Diabetes 2001;50(suppl 1):S160–S163.")
44
Insulin Sensitivität Beta-Zell Funktion
Belfast Diet Study Reduzierte ß-Zell-Funktion bei stabiler Insulin Sensitivität in Typ-2-Diabetes Insulin Sensitivität 60 Beta-Zell Funktion 80 60 40 40 HOMA % Sensitivität HOMA % Beta 20 20 2 4 6 2 4 6 Jahre ab Diagnose Jahre ab Diagnose Data from the first six years of 10-year follow-up of the Belfast Diet Study: Data from 67 newly diagnosed subjects with type 2 diabetes mellitus (N=432) who required oral antihyperglycemic therapy or insulin due to secondary failure of diet therapy during years 5 to 7 HOMA=Homeostasis Model Assessment; data expressed as percentages of values in lean nondiabetic reference population Adapted from Levy J et al Diabet Med 1998;15:290–296.
 who required oral antihyperglycemic therapy or insulin due to secondary failure of diet therapy during years 5 to 7. HOMA=Homeostasis Model Assessment; data expressed as percentages of values in lean nondiabetic reference population. Adapted from Levy J et al Diabet Med 1998;15:290–296.")
45
GLP-1 und GIP als wichtige Inkretine
Synthese in L-Zellen im distalen Darm (Ileum und Colon) Stimuliert Glucose-abhängige Insulin- freisetzung Synthese in K-Zellen im proximalen Darm (Duodenum) Stimuliert Glucose-abhängige Insulinfreisetzung Andere Effekte Hemmung der hepatischen Glukoseproduktion durch Hemmung der Glukagonsekretion (glukoseabhängig) Hemmung der Magenentleerung, Reduktion von Nahrungsaufnahme & Körpergewicht Stimulation der Betazellproliferation & des Betazellüberlebens (Tiermodelle & isolierte humane Inseln) Minimale Effekte auf die Magenentleerung, keine signifikanten Effekte auf Sättigung & Körpergewicht Potentiell Stimulation der Betazellproliferation & des Betazellüberlebens (isolierte Inselzelllinien) Drucker DJ. Diabetes Care. 2003;26:2929–2940; Ahrén B. Curr Diab Rep. 2003;3:365–372; Drucker DJ. Gastroenterology. 2002;122: 531–544; Farilla L et al. Endocrinology. 2003;144:5149–5158; Trümper A et al. Mol Endocrinol. 2001;15:1559–1570; Trümper A et al. J Endocrinol. 2002;174:233–246; Wideman RD et al. Horm Metab Res. 2004;36:782–786. References: 1. Drucker DJ. Enhancing incretin action for the treatment of type 2 diabetes. Diabetes Care. 2003;26:2929–2940. 2. Ahrén B. Gut peptides and type 2 diabetes mellitus treatment. Curr Diab Rep. 2003;3:365–372. 3. Drucker DJ. Biological actions and therapeutic potential of the glucagon-like peptides. Gastroenterology. 2002;122:531–544. 4. Nauck MA, Niedereichholz U, Ettler R, et al. Glucagon-like peptide 1 inhibition of gastric emptying outweighs its insulinotropic effects in healthy humans. Am J Physiol. 1997;273:E981–E988. 5. Farilla L, Bulotta A, Hirshberg B, et al. Glucagon-like peptide 1 inhibits cell apoptosis and improves glucose responsiveness of freshly isolated human islets. Endocrinology. 2003;144:5149–5158. 6. Farilla L, Hui H, Bertolotto C, et al. Glucagon-like peptide-1 promotes islet cell growth and inhibits apoptosis in Zucker diabetic rats. Endocrinology. 2002;143:4397–4408. 7. Meier JJ, Goetze O, Anstipp J, et al. Gastric inhibitory polypeptide does not inhibit gastric emptying in humans. Am J Physiol Endocrinol Metab. 2004;286:E621–E625. 8. Nauck MA, Heimesaat MM, Ørskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved incretin activity of glucagon- like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest. 1993;91:301–307. 9. Trümper A, Trümper K, Trusheim H, Arnold R, Göke B, Hörsch D. Glucose-dependent insulinotropic polypeptide is a growth factor for beta (INS-1) cells by pleiotropic signaling. Mol Endocrinol. 2001;15:1559–1570. 10. Trümper A, Trümper K, Hörsch D. Mechanisms of mitogenic and anti-apoptotic signaling by glucose-dependent insulinotropic polypeptide in β(INS-1)-cells. J Endocrinol. 2002;174:233–246.
 Stimuliert Glucose-abhängige Insulin- freisetzung. Synthese in K-Zellen im proximalen Darm (Duodenum) Stimuliert Glucose-abhängige Insulinfreisetzung. Andere Effekte. Hemmung der hepatischen Glukoseproduktion durch Hemmung der Glukagonsekretion (glukoseabhängig) Hemmung der Magenentleerung, Reduktion von Nahrungsaufnahme & Körpergewicht. Stimulation der Betazellproliferation & des Betazellüberlebens (Tiermodelle & isolierte humane Inseln) Minimale Effekte auf die Magenentleerung, keine signifikanten Effekte auf Sättigung & Körpergewicht. Potentiell Stimulation der Betazellproliferation & des Betazellüberlebens (isolierte Inselzelllinien) Drucker DJ. Diabetes Care. 2003;26:2929–2940; Ahrén B. Curr Diab Rep. 2003;3:365–372; Drucker DJ. Gastroenterology. 2002;122: 531–544; Farilla L et al. Endocrinology. 2003;144:5149–5158; Trümper A et al. Mol Endocrinol. 2001;15:1559–1570; Trümper A et al. J Endocrinol. 2002;174:233–246; Wideman RD et al. Horm Metab Res. 2004;36:782–786. References: 1. Drucker DJ. Enhancing incretin action for the treatment of type 2 diabetes. Diabetes Care. 2003;26:2929– Ahrén B. Gut peptides and type 2 diabetes mellitus treatment. Curr Diab Rep. 2003;3:365– Drucker DJ. Biological actions and therapeutic potential of the glucagon-like peptides. Gastroenterology. 2002;122:531– Nauck MA, Niedereichholz U, Ettler R, et al. Glucagon-like peptide 1 inhibition of gastric emptying outweighs its insulinotropic effects in healthy humans. Am J Physiol. 1997;273:E981–E Farilla L, Bulotta A, Hirshberg B, et al. Glucagon-like peptide 1 inhibits cell apoptosis and improves glucose responsiveness of freshly isolated human islets. Endocrinology. 2003;144:5149– Farilla L, Hui H, Bertolotto C, et al. Glucagon-like peptide-1 promotes islet cell growth and inhibits apoptosis in Zucker diabetic rats. Endocrinology. 2002;143:4397– Meier JJ, Goetze O, Anstipp J, et al. Gastric inhibitory polypeptide does not inhibit gastric emptying in humans. Am J Physiol Endocrinol Metab. 2004;286:E621–E Nauck MA, Heimesaat MM, Ørskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved incretin activity of glucagon- like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest. 1993;91:301– Trümper A, Trümper K, Trusheim H, Arnold R, Göke B, Hörsch D. Glucose-dependent insulinotropic polypeptide is a growth factor for beta (INS-1) cells by pleiotropic signaling. Mol Endocrinol. 2001;15:1559– Trümper A, Trümper K, Hörsch D. Mechanisms of mitogenic and anti-apoptotic signaling by glucose-dependent insulinotropic polypeptide in β(INS-1)-cells. J Endocrinol. 2002;174:233–246.")
46
Verminderter Inkretineffekt bei Typ 2 Diabetes
Healthy controls (n=8) Type 2 diabetes (n=14) Oral glucose (50 g/400 ml) Isoglycemic intravenous glucose 6/Nauck 1986, p 47, C2, ¶2, L4-5; p 48, Fig 1, bottom 2 panels; p 49, Fig 2, top 2 panels 80 Normal incretin effect 80 60 60 Diminished incretin effect IR insulin (mU/L) 40 IR insulin (mU/L) 40 * 1/Vilsbøll 2004, p 357, C2, ¶1, L3-10 2/Brubaker, p 2653, C1, ¶2, L1-6, C2, L1-7 3/Holst 2004, p E199, C1, ¶1, L1-5 2/Brubaker, p 2653, C2, L7-10 4/Nauck 1986, p 46, C2, ¶2, L1-3; p 51, C2, ¶2, L1-5 p 48, C2, ¶4, L4-6 p 48, C2, ¶5, L3-6; p 49, C1, L7-8, C2, L1-3 20 * 20 An oral glucose load results in a greater insulin response than the response caused by an intravenous glucose load matched to produce a similar glycemic profile. This phenomenon is termed “the incretin effect” because it is attributable to the release of incretin hormones that occurs after oral but not intravenous administration of glucose.1-3 Studies in humans and animal models have shown that the incretin hormones GLP-1 and GIP are believed to account for almost the entire incretin effect that facilitates disposal of ingested nutrients.2 A clinical study showed that the incretin effect was diminished in patients with type 2 diabetes (n=14) compared with metabolically healthy control subjects (n=8).4 As shown on the graphs, glucose profiles were closely mimicked at similar levels after oral versus intravenous glucose administration.4 This matching of intravenous glucose loads to oral glucose loads after ingestion was achieved in both control subjects and patients with diabetes.4 Beta-cell secretory responses, reflected by increases in plasma levels of immunoreactive (IR) insulin, are shown on the bottom graphs in the slide. These graphs show that plasma IR insulin peaks were delayed and diminished in patients with type 2 diabetes. Although insulin levels were greater after oral glucose ingestion versus intravenous administration in both groups, the incretin effects were markedly less pronounced in diabetic patients.4 –10 –5 60 120 180 –10 –5 60 120 180 Time (min) Time (min) Incretin Levels Incretin Actions GLP-1 Decreased Intact GIP *p0.05 vs. respective value after oral load IR=immunoreactive Adapted from Nauck M et al Diabetologia 1986;29:46–52. References Vilsbøll T, Holst JJ. Incretins, insulin secretion and type 2 diabetes mellitus. Diabetologia 2004;47:357–366. Brubaker PL, Drucker DJ. Minireview: Glucagon-like peptides regulate cell proliferation and apoptosis in the pancreas, gut, and central nervous system. Endocrinology 2004;145:2653–2659. Holst JJ, Gromada J. Role of incretin hormones in the regulation of insulin secretion in diabetic and nondiabetic humans. Am J Physiol Endocrinol Metab 2004;287:E199–E206. Nauck M, Stöckmann F, Ebert R et al. Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia 1986;29:46–52.
 Type 2 diabetes (n=14) Oral glucose (50 g/400 ml) Isoglycemic intravenous glucose. 6/Nauck 1986, p 47, C2, ¶2, L4-5; p 48, Fig 1, bottom 2 panels; p 49, Fig 2, top 2 panels. 80. Normal incretin effect Diminished incretin effect. IR insulin (mU/L) 40. IR insulin (mU/L) 40. * 1/Vilsbøll 2004, p 357, C2, ¶1, L /Brubaker, p 2653, C1, ¶2, L1-6, C2, L1-7. 3/Holst 2004, p E199, C1, ¶1, L1-5. 2/Brubaker, p 2653, C2, L /Nauck 1986, p 46, C2, ¶2, L1-3; p 51, C2, ¶2, L1-5. p 48, C2, ¶4, L4-6. p 48, C2, ¶5, L3-6; p 49, C1, L7-8, C2, L * 20. An oral glucose load results in a greater insulin response than the response caused by an intravenous glucose load matched to produce a similar glycemic profile. This phenomenon is termed the incretin effect because it is attributable to the release of incretin hormones that occurs after oral but not intravenous administration of glucose.1-3. Studies in humans and animal models have shown that the incretin hormones GLP-1 and GIP are believed to account for almost the entire incretin effect that facilitates disposal of ingested nutrients.2. A clinical study showed that the incretin effect was diminished in patients with type 2 diabetes (n=14) compared with metabolically healthy control subjects (n=8).4. As shown on the graphs, glucose profiles were closely mimicked at similar levels after oral versus intravenous glucose administration.4 This matching of intravenous glucose loads to oral glucose loads after ingestion was achieved in both control subjects and patients with diabetes.4. Beta-cell secretory responses, reflected by increases in plasma levels of immunoreactive (IR) insulin, are shown on the bottom graphs in the slide. These graphs show that plasma IR insulin peaks were delayed and diminished in patients with type 2 diabetes. Although insulin levels were greater after oral glucose ingestion versus intravenous administration in both groups, the incretin effects were markedly less pronounced in diabetic patients.4. –10. – –10. – Time (min) Time (min) Incretin Levels. Incretin Actions. GLP-1. Decreased. Intact. GIP. *p0.05 vs. respective value after oral load. IR=immunoreactive. Adapted from Nauck M et al Diabetologia 1986;29:46–52. References. Vilsbøll T, Holst JJ. Incretins, insulin secretion and type 2 diabetes mellitus. Diabetologia 2004;47:357–366. Brubaker PL, Drucker DJ. Minireview: Glucagon-like peptides regulate cell proliferation and apoptosis in the pancreas, gut, and central nervous system. Endocrinology 2004;145:2653–2659. Holst JJ, Gromada J. Role of incretin hormones in the regulation of insulin secretion in diabetic and nondiabetic humans. Am J Physiol Endocrinol Metab 2004;287:E199–E206. Nauck M, Stöckmann F, Ebert R et al. Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia 1986;29:46–52.")
47
GLP-1-Spiegel bei Typ-2-Diabetes erniedrigt
Meal Test Study GLP-1-Spiegel bei Typ-2-Diabetes erniedrigt 20 NGT (n=33) Typ-2-Diabetes (n=54) * * 15 GLP-1 (pmol/L) 10 5 60 120 180 240 Zeit (Minuten) *p<0.05, type 2 diabetes vs. NGT Meal started at time 0 and finished at 10–15 minutes. Adapted from Toft-Nielsen M-B et al J Clin Endocrinol Metab 2001;86:3717–3723.
 Typ-2-Diabetes (n=54) * * 15. GLP-1 (pmol/L) Zeit (Minuten) *p<0.05, type 2 diabetes vs. NGT. Meal started at time 0 and finished at 10–15 minutes. Adapted from Toft-Nielsen M-B et al J Clin Endocrinol Metab 2001;86:3717–3723.")
48
Präklinik GLP-1 erhöhte die Proliferation und hemmte die Apoptose von ß-Zellen in Zucker Diabetic Rats ß-Zell Proliferation ß-Zell Apoptose 2.5 30 25 2.0 1.4-facher Anstieg (p<0.05) 20 1.5 3.6-fache Abnahme (p<0.001) proliferierende ß-Zellen (%) apoptotische ß-Zellen (%) 15 1.0 10 0.5 5 GLP-1 Behandlung GLP-1 Behandlung Kontrolle Kontrolle Study in Zucker diabetic rats that received a two-day infusion of GLP-1 or saline followed by a glucose tolerance test. Pancreatic sections were drawn to measure islet mass, β-cell proliferation, and apoptosis. Adapted from Farilla L et al Endocrinology 2002;143:4397–4408.
 fache Abnahme (p<0.001) proliferierende ß-Zellen (%) apoptotische ß-Zellen (%) GLP-1 Behandlung. GLP-1 Behandlung. Kontrolle. Kontrolle. Study in Zucker diabetic rats that received a two-day infusion of GLP-1 or saline followed by a glucose tolerance test. Pancreatic sections were drawn to measure islet mass, β-cell proliferation, and apoptosis. Adapted from Farilla L et al Endocrinology 2002;143:4397–4408.")
49
GLP-1 verlängert die Überlebenszeit von humanen Langerhans` Inseln In Vitro
1/Farilla 2003, p 5151, C2, ¶2, L11-13, Fig 1 Kontrolle GLP-1–behandelte Inseln Unter Behandlung mit GLP-1 verlängerte sich die Überlebenszeit humaner Langerhans` Inseln in vitro Day 1 An in vitro study was conducted to evaluate the potential of GLP-1 to improve the viability and function of freshly isolated human islets. Islets were maintained in culture for five days in the presence or absence of GLP-1.1 During the five days of culture, islets in the control group showed important morphologic changes. At day 1, islets maintained their physiologic spherical shape (panel A). By day 3, many islets showed a progressive loss of structure, losing the acellular membrane that surrounds them (panel C). At day 5, the three-dimensional structure of many cell aggregates had deteriorated to a two-dimensional structure typical of a cell monolayer (panel E). In contrast, islets treated with GLP-1 retained their three-dimensional organization for a longer period of time (panels B, D, and F). Overall, the number of islets with a preserved three-dimensional structure by day 5 had declined by 45% in the control group versus a 15% reduction in the GLP-1–treated islets (p<0.01).1 Additional analyses showed that the number of apoptotic cells was significantly lower in GLP-1–treated islets versus control islets at day 3 (6.1% vs. 15.5%, respectively; p<0.01) and day 5 (8.9% vs. 18.9%, respectively; p<0.01), and that intracellular insulin content was markedly enhanced in islets cultured with GLP-1 versus control (p<0.001 at day 5).1 These results help to provide evidence that GLP-1 preserves morphology and function and inhibits apoptosis of islet cells.1 1/Farilla 2003, p 5150, C1, ¶1, L4-6, ¶3, L1-2, 6-10 p 5151, C2, ¶2, L1-13, Fig 1; p 5152, C1, L2-5 p 5153, C1, ¶1, L10-13, C2, L1-2; p 5154, C2, ¶2, L5-8 p 5157, C2, ¶1, L4-8, 12-14; p 5158, C1, ¶1, L1-3 Day 3 Day 5 Adapted from Farilla L et al Endocrinology 2003;144:5149–5158. Reference 1. Farilla L, Bulotta A, Hirshberg B et al. Glucagon-like peptide inhibits cell apoptosis and improves glucose responsiveness of freshly isolated human islets. Endocrinology 2003;144:5149–5158.
. By day 3, many islets showed a progressive loss of structure, losing the acellular membrane that surrounds them (panel C). At day 5, the three-dimensional structure of many cell aggregates had deteriorated to a two-dimensional structure typical of a cell monolayer (panel E). In contrast, islets treated with GLP-1 retained their three-dimensional organization for a longer period of time (panels B, D, and F). Overall, the number of islets with a preserved three-dimensional structure by day 5 had declined by 45% in the control group versus a 15% reduction in the GLP-1–treated islets (p<0.01).1. Additional analyses showed that the number of apoptotic cells was significantly lower in GLP-1–treated islets versus control islets at day 3 (6.1% vs. 15.5%, respectively; p<0.01) and day 5 (8.9% vs. 18.9%, respectively; p<0.01), and that intracellular insulin content was markedly enhanced in islets cultured with GLP-1 versus control (p<0.001 at day 5).1. These results help to provide evidence that GLP-1 preserves morphology and function and inhibits apoptosis of islet cells.1. 1/Farilla 2003, p 5150, C1, ¶1, L4-6, ¶3, L1-2, p 5151, C2, ¶2, L1-13, Fig 1; p 5152, C1, L2-5. p 5153, C1, ¶1, L10-13, C2, L1-2; p 5154, C2, ¶2, L5-8. p 5157, C2, ¶1, L4-8, 12-14; p 5158, C1, ¶1, L1-3. Day 3. Day 5. Adapted from Farilla L et al Endocrinology 2003;144:5149–5158. Reference. 1. Farilla L, Bulotta A, Hirshberg B et al. Glucagon-like peptide inhibits cell apoptosis and improves glucose responsiveness of freshly isolated human islets. Endocrinology 2003;144:5149–5158.")
50
Wirkung der Inkretinhormone GLP-1 und GIP
Nahrungs-aufnahme glucoseabhängig Insulin von Beta Zellen (GLP-1 und GIP) Insulin erhöht die periphere Glucose- aufnahme Darmtrakt Pankreas Freigabe von Inkretin Beta Zellen Blut Glucosekontrolle Alpha Zellen aktives GLP-1 and GIP Erhöhtes Insulin und veringertes Glucagon reduziert die hepatische Glucosefreigabe Glucagon von Alpha Zellen (GLP-1) glucoseabhänhig Adapted from Brubaker PL, Drucker DJ Endocrinology 2004;145:2653–2659; Zander M et al Lancet 2002;359:824–830; Ahrén B Curr Diab Rep 2003;3:365–372; Buse JB et al. In Williams Textbook of Endocrinology. 10th ed. Philadelphia, Saunders, 2003:1427–1483.
 Insulin erhöht die periphere Glucose- aufnahme. Darmtrakt. Pankreas. Freigabe von Inkretin. Beta Zellen. Blut Glucosekontrolle. Alpha Zellen. aktives GLP-1 and GIP. Erhöhtes Insulin und veringertes Glucagon reduziert die hepatische Glucosefreigabe. Glucagon. von Alpha Zellen (GLP-1) glucoseabhänhig. Adapted from Brubaker PL, Drucker DJ Endocrinology 2004;145:2653–2659; Zander M et al Lancet 2002;359:824–830; Ahrén B Curr Diab Rep 2003;3:365–372; Buse JB et al. In Williams Textbook of Endocrinology. 10th ed. Philadelphia, Saunders, 2003:1427–1483.")
51
Rascher enzymatischer Abbau der Inkretinhormone durch Dipeptidyl Peptidase 4 (DPP-4)
3/Rasmussen, p 20, Fig. 1a,b + legend 1/Evans, p 577, C1, ¶1, L1-6, C1, ¶2, L1-7 2/Drucker 2003A, p 89, C2, ¶1, L9-12,20-24, ¶2, L1-6 Cell membrane Cytosol N C 1/Evans, p 577, C1, ¶1, L1-6 2/Drucker 2003A, p 89, C2, ¶1, L9-12, ¶2, L1-6 p 577, C1, ¶2, L1-7 p 89, C2, ¶1, L20-24 3/Rasmussen, p 19, C1, ¶1, L1-5 p 90, Box 1 p 19, C2, ¶3, L8-9; p 20, C1, L1-3, Fig 1a,b; p 23, C1, ¶1, L1-5, ¶2, L1-5; p 19, C2, ¶2, L2-4 p 577, C2, ¶1, L23-25 p 92, C2, ¶2, L1-11 Dipeptidyl peptidase 4 (DPP-4) is a membrane-bound serine protease of the prolyl oligopeptidase enzyme family.1 DPP-4 exists in two forms. In its membrane-bound form, it is widely expressed on cells in most tissues.2 In its soluble, circulating form, DPP-4 is found in the blood, saliva, urine, and synovial fluid.1,2 DPP-4 modulates the biological activity of several peptide hormones, chemokines, and neuropeptides by specifically cleaving from the N-terminus after a proline or alanine.3 The slide shows the structure of the extracellular region of DPP-4. The catalytic (active) site is located in a small pocket within a large cavity formed between the alpha/beta-hydrolase domain and an eight-bladed beta-propeller domain. Residues from both domains participate in inhibitor binding. The structure of the active site reveals why most natural DPP-4 substrates are short peptides (<80 amino acids) such as GLP-1 and GIP: only elongated peptides or unfolded or partly unfolded protein fragments can reach the site.2,3 The enzymatic activity of DPP-4 on incretins, especially GLP-1, suggests a pharmacologic role for DPP-4 inhibitors in the treatment of patients with type 2 diabetes.1,2 DPP-4 ist eine Serinprotease der Prolyl Oligopeptidase Enzym Familie, die in 2 Formen vorkommt: Membrangebunden Im Plasma gelöst Adapted from Evans DM IDrugs 2002;5:577–585; Drucker DJ Expert Opin Investig Drugs 2003;12:87–100; Rasmussen HB et al Nat Struct Biol 2003;10:19–25. References Evans DM. Dipeptidyl peptidase IV inhibitors. IDrugs 2002;5:577–585. Drucker DJ. Therapeutic potential of dipeptidyl peptidase IV inhibitors for the treatment of type 2 diabetes. Expert Opin Investig Drugs 2003;12:87–100. Rasmussen HB, Branner S, Wiberg FC et al. Crystal structure of human dipeptidyl peptidase IV/CD26 in complex with a substrate analog. Nat Struct Biol 2003;10:19–25.
 is a membrane-bound serine protease of the prolyl oligopeptidase enzyme family.1 DPP-4 exists in two forms. In its membrane-bound form, it is widely expressed on cells in most tissues.2 In its soluble, circulating form, DPP-4 is found in the blood, saliva, urine, and synovial fluid.1,2 DPP-4 modulates the biological activity of several peptide hormones, chemokines, and neuropeptides by specifically cleaving from the N-terminus after a proline or alanine.3. The slide shows the structure of the extracellular region of DPP-4. The catalytic (active) site is located in a small pocket within a large cavity formed between the alpha/beta-hydrolase domain and an eight-bladed beta-propeller domain. Residues from both domains participate in inhibitor binding. The structure of the active site reveals why most natural DPP-4 substrates are short peptides (<80 amino acids) such as GLP-1 and GIP: only elongated peptides or unfolded or partly unfolded protein fragments can reach the site.2,3. The enzymatic activity of DPP-4 on incretins, especially GLP-1, suggests a pharmacologic role for DPP-4 inhibitors in the treatment of patients with type 2 diabetes.1,2. DPP-4 ist eine Serinprotease der Prolyl Oligopeptidase Enzym Familie, die in 2 Formen vorkommt: Membrangebunden. Im Plasma gelöst. Adapted from Evans DM IDrugs 2002;5:577–585; Drucker DJ Expert Opin Investig Drugs 2003;12:87–100; Rasmussen HB et al Nat Struct Biol 2003;10:19–25. References. Evans DM. Dipeptidyl peptidase IV inhibitors. IDrugs 2002;5:577–585. Drucker DJ. Therapeutic potential of dipeptidyl peptidase IV inhibitors for the treatment of type 2 diabetes. Expert Opin Investig Drugs 2003;12:87–100. Rasmussen HB, Branner S, Wiberg FC et al. Crystal structure of human dipeptidyl peptidase IV/CD26 in complex with a substrate analog. Nat Struct Biol 2003;10:19–25.")
52
Intestinale GIP und GLP-1 Freigabe
DPP-4-Hemmung erhöht die Spiegel der biologisch aktiven Inkretine GLP-1 und GIP Mahlzeit DPP-4 inhibitor DPP-4 Enzym Intestinale GIP und GLP-1 Freigabe GIP (1–42) GLP-1 (7–36) GIP (3–42) GLP-1 (9–36) Schneller Abbau (Minuten) GIP und GLP-1 Wirkungen Adapted from Deacon CF et al Diabetes 1995;44:1126–1131; Kieffer TJ et al Endocrinology 1995;136:3585–3596; Ahrén B Curr Diab Rep 2003;3:365–372; Deacon CF et al J Clin Endocrinol Metab 1995;80:952–957; Weber AE J Med Chem 2004;47:4135–4141.
 GLP-1 (7–36) GIP (3–42) GLP-1 (9–36) Schneller Abbau (Minuten) GIP und GLP-1 Wirkungen. Adapted from Deacon CF et al Diabetes 1995;44:1126–1131; Kieffer TJ et al Endocrinology 1995;136:3585–3596; Ahrén B Curr Diab Rep 2003;3:365–372; Deacon CF et al J Clin Endocrinol Metab 1995;80:952–957; Weber AE J Med Chem 2004;47:4135–4141.")
53
DPP-4-Hemmung erhöht im Tierversuch Insulingehalt
und Masse der Betazellen HFD/STZ diabetic mice were treated with vehicle, des-fluoro-sitagliptin (des-F-sitagliptin), or glipizide. Pancreatic sections from various groups of HFD/STZ mice and nondiabetic control mice were stained with anti-insulin antibody or anti-glucagon antibody. (A) Shown are images with the overlay of the insulin (green) and glucagon (red) staining HFD/STZ diabetic mice were treated with vehicle, des-fluoro-sitagliptin (des-F-sitagliptin), or glipizide. Pancreatic sections from various groups of HFD/STZ mice and nondiabetic control mice were stained with anti-insulin antibody or anti-glucagon antibody. (A) Shown are images with the overlay of the insulin (green) and glucagon (red) staining (B) Insulin-positive -cell-to-total islet area ratio was also measured from these images. (C) Pancreata from separated groups of animals of this study were collected, weighed, and used to measure insulin content after acid ethanol extraction. *P < 0.01 vs. the vehicle-treated group, n = 40 islets in each group. Mu J et al., Diabetes 55: , 2006
, or glipizide. Pancreatic sections from various groups of HFD/STZ mice and nondiabetic control mice were stained with anti-insulin antibody or anti-glucagon antibody. (A) Shown are images with the overlay of the insulin (green) and glucagon (red) staining. HFD/STZ diabetic mice were treated with vehicle, des-fluoro-sitagliptin (des-F-sitagliptin), or glipizide. Pancreatic sections from various groups of HFD/STZ mice and nondiabetic control mice were stained with anti-insulin antibody or anti-glucagon antibody. (A) Shown are images with the overlay of the insulin (green) and glucagon (red) staining. (B) Insulin-positive -cell-to-total islet area ratio was also measured from these images. (C) Pancreata from separated groups of animals of this study were collected, weighed, and used to measure insulin content after acid ethanol extraction. *P < 0.01 vs. the vehicle-treated group, n = 40 islets in each group. Mu J et al., Diabetes 55: ,")
54
Schmerzen bei diabetischer Polyneuropathie

55
Neuropathische Schmerzen
Ursache ist eine Schädigung Schmerz leitender oder Schmerz verarbeitender Systeme im peripheren oder zentralen Nervensystem1 Neuropathischer Schmerz: Endprodukt einer veränderten peripheren, spinalen und supraspinalen Signalverarbeitung2 Periphere Neuropathie: Die Terminologie legt fest, dass am Anfang eines neuropathischen Schmerzgeschehens immer eine primäre Noxe am peripheren Nerv besteht, die erst im weiteren Verlauf zu sekundären Veränderungen im Rückenmark führen kann. Diese im ZNS gelegenen Veränderungen können dann einen wesentlichen Anteil am Unterhalt des Schmerzgeschehens haben. Aufgrund seines Entstehungsmechanismus wird der Schmerz jedoch als peripherer neuropathischer Schmerz bezeichnet (im Gegensatz zum zentralen neuropathischen Schmerz, hier liegt die primäre Noxe im ZNS). Es gibt einen fokalen Befall oder es sind diffus mehrere Nerven betroffen (= Polyneuropathie). 1. Leitlinie Neuropathischer Schmerz der DGN (Deutsche Gesellschaft Neurologie) 2005 (in press) 2. Tölle TR, Baron R Fortschritte der Medizin 120.Jg.-Originalien Nr. II-III/2002, 49-59
. Es gibt einen fokalen Befall oder es sind diffus mehrere Nerven betroffen (= Polyneuropathie). 1. Leitlinie Neuropathischer Schmerz der DGN (Deutsche Gesellschaft Neurologie) 2005 (in press) 2. Tölle TR, Baron R Fortschritte der Medizin 120.Jg.-Originalien Nr. II-III/2002,")
56
Schmerzwahrnehmung – neuronale Strukturen
Kortex und andere Hirnstrukturen Thalamus Mesenzephalon (zentrales Höhlengrau) Medulla oblongata (Raphekerne) Hinterhorn des Rückenmarks (Substantia gelatinosa) Primäres sensorisches Neuron Mit diesem Schema sollen einige als Grundlage zur Pathophysiologie des neuropathischen Schmerzes bekannte Mechanismen exemplarisch aufgezeichnet werden. Für die Wahrnehmung von Schmerz verfügen Menschen über spezielle, anatomisch und physikalisch gut charakterisierte Strukturen: Die erste strukturelle Einheit ist ein sensorisches Neuron in der Peripherie, das Rezeptoren für die Schmerzreize enthält. Vom afferenten Schmerzneuron wird die Meldung ‚Schmerz‘ zum Rückenmark geleitet. Dort erfolgt unter dem Einfluss umfangreicher synaptischer Modulation eine Umschaltung auf Neurone, die in supraspinale Hirnregionen projizieren. Auf dem Weg von der Schmerzauslösung in der Peripherie bis zur Schmerzverarbeitung im Kortex sind zahlreiche Mediatoren und Modulatoren beteiligt, die im Zusammenwirken darüber entscheiden, ob und wie ein eingehender Reiz im somatischen Kortex als schmerzhaft empfunden wird oder nicht. Sehr unterschiedliche pathophysiologische Vorgänge können ein klinisch gleiches Bild verursachen, andererseits können verschiedene klinische Krankheitszustände den gleichen Verletzungsmechanismus am Nervensystem auslösen. Klingelzugmodell des Schmerzes von Descartes Hiemke C, in Handbuch Chronischer Schmerz, Egle UT et al., Schattauer 2003
 Medulla oblongata (Raphekerne) Hinterhorn des Rückenmarks (Substantia gelatinosa) Primäres sensorisches Neuron. Mit diesem Schema sollen einige als Grundlage zur Pathophysiologie des neuropathischen Schmerzes bekannte Mechanismen exemplarisch aufgezeichnet werden. Für die Wahrnehmung von Schmerz verfügen Menschen über spezielle, anatomisch und physikalisch gut charakterisierte Strukturen: Die erste strukturelle Einheit ist ein sensorisches Neuron in der Peripherie, das Rezeptoren für die Schmerzreize enthält. Vom afferenten Schmerzneuron wird die Meldung ‚Schmerz‘ zum Rückenmark geleitet. Dort erfolgt unter dem Einfluss umfangreicher synaptischer Modulation eine Umschaltung auf Neurone, die in supraspinale Hirnregionen projizieren. Auf dem Weg von der Schmerzauslösung in der Peripherie bis zur Schmerzverarbeitung im Kortex sind zahlreiche Mediatoren und Modulatoren beteiligt, die im Zusammenwirken darüber entscheiden, ob und wie ein eingehender Reiz im somatischen Kortex als schmerzhaft empfunden wird oder nicht. Sehr unterschiedliche pathophysiologische Vorgänge können ein klinisch gleiches Bild verursachen, andererseits können verschiedene klinische Krankheitszustände den gleichen Verletzungsmechanismus am Nervensystem auslösen. Klingelzugmodell des Schmerzes von Descartes. Hiemke C, in Handbuch Chronischer Schmerz, Egle UT et al., Schattauer")
57
afferente Schmerzfasern
Modulation der Schmerzwahrnehmung im Rückenmark Glycin* Serotonin* Noradrenalin* GABA* *Absteigende Bahnen Hinterhorn- neuron *Lokale Interneurone Substanz P Neurokinin A CGRP Glutamat Somatostatin Prinzip der Schmerzwahrnehmung: Über afferente Schmerzfasern (C-Fasern, A-Fasern) eingehende Reize aktivieren ein Hinterhornneuron, indem sie Glutamat, Substanz P, Neurokinin A oder Calcitonin Gen Related Peptide (CGRP) freisetzen. Die Weiterleitung des Schmerzreizes wird gehemmt/moduliert durch: ebenfalls aus einer afferenten Faser freigesetztes Somatostatin; lokale Interneurone, die -Amino-Buttersäure (GABA), Enkephalin oder Glycin enthalten; absteigende Bahnen mit Serotonin und Noradrenalin als Transmitter. Pathophysiologie des neuropathischen Schmerzes [Ludwig & Baron, DNP, 5/2004]: In Folge der Schädigung peripherer Neurone kommt es zu einer Sensibilisierung und Überempfindlichkeit peripherer C-Faser-Afferenzen. Chronisch sensibilisierte Nozizeptoren bilden eine Ruheaktivität aus, besitzen eine erniedrigte Schwelle gegenüber noxischen Reizen und erzeugen eine supranormale Anwort auf überschwellige Reize. Zudem können geschädigte primär afferente C-Nozizeptoren durch Expression von Na-Kanälen ektope Nervenimpulse generieren. Die ständige C-Faser-Aktivität resultiert über eine Ausschüttung von Glutamat und Substanz P an den zentralen Endigungen im Hinterhorn in einer Aktivierung der WDR (Wide-dynamic-range) Neurone, die über second-messenger-Prozesse zu einer Veränderung der Gen-expression und somit zu einer zentralen Sensibilisierung führen. Weiterhin kommt es zu einer Degeneration inhibitorischer Systeme: Durch chronisch nozizeptive Aktivität kommt es zum einen zu einem Funktionsverlust absteigender hemmender noradrenerger und serotonerger Bahnen, zum anderen zur Degeneration GABAerger Interneurone im Hinterhorn. Durch die Plastizität des nozizeptiven Systems können diese Mechanismen zur Schmerzchronifizierung nicht nur in der Peripherie und im Rückenmark, sondern auch in den suprasspinalen Schmerz verarbeitenden Systemen (z.B. Thalamus, somatosensorischer Cortex) vorkommen. Enkephalin* CGRP = Calcitonin Gen Related Peptide afferente Schmerzfasern Hiemke C, in Handbuch Chronischer Schmerz, Egle UT et al., Schattauer 2003
 eingehende Reize aktivieren ein Hinterhornneuron, indem sie Glutamat, Substanz P, Neurokinin A oder Calcitonin Gen Related Peptide (CGRP) freisetzen. Die Weiterleitung des Schmerzreizes wird gehemmt/moduliert durch: ebenfalls aus einer afferenten Faser freigesetztes Somatostatin; lokale Interneurone, die -Amino-Buttersäure (GABA), Enkephalin oder Glycin enthalten; absteigende Bahnen mit Serotonin und Noradrenalin als Transmitter. Pathophysiologie des neuropathischen Schmerzes [Ludwig & Baron, DNP, 5/2004]: In Folge der Schädigung peripherer Neurone kommt es zu einer Sensibilisierung und Überempfindlichkeit peripherer C-Faser-Afferenzen. Chronisch sensibilisierte Nozizeptoren bilden eine Ruheaktivität aus, besitzen eine erniedrigte Schwelle gegenüber noxischen Reizen und erzeugen eine supranormale Anwort auf überschwellige Reize. Zudem können geschädigte primär afferente C-Nozizeptoren durch Expression von Na-Kanälen ektope Nervenimpulse generieren. Die ständige C-Faser-Aktivität resultiert über eine Ausschüttung von Glutamat und Substanz P an den zentralen Endigungen im Hinterhorn in einer Aktivierung der WDR (Wide-dynamic-range) Neurone, die über second-messenger-Prozesse zu einer Veränderung der Gen-expression und somit zu einer zentralen Sensibilisierung führen. Weiterhin kommt es zu einer Degeneration inhibitorischer Systeme: Durch chronisch nozizeptive Aktivität kommt es zum einen zu einem Funktionsverlust absteigender hemmender noradrenerger und serotonerger Bahnen, zum anderen zur Degeneration GABAerger Interneurone im Hinterhorn. Durch die Plastizität des nozizeptiven Systems können diese Mechanismen zur Schmerzchronifizierung nicht nur in der Peripherie und im Rückenmark, sondern auch in den suprasspinalen Schmerz verarbeitenden Systemen (z.B. Thalamus, somatosensorischer Cortex) vorkommen. Enkephalin* CGRP = Calcitonin Gen Related Peptide. afferente Schmerzfasern. Hiemke C, in Handbuch Chronischer Schmerz, Egle UT et al., Schattauer")
58
Schmerzmodulation – Serotonin (5-HT) und Noradrenalin (NA)
Absteigende Modulation PAG kontrolliert indirekt die Schmerzleitung in die Columna dorsalis Neuropathische Schmerzen sind mit vermehrter Erregbarkeit und verminderter Hemmung von aufsteigenden Schmerzbahnen assoziiert Absteigende Bahnen modulieren aufsteigende Signale NA und 5-HT sind wichtige Neurotransmitter der absteigenden hemmenden Schmerzbahnen Die gesteigerte Verfügbarkeit von NA und 5- HT kann die Schmerzhemmung zentral fördern ACC T H Amygdala PAG DLPT Aufsteigende Schmerzbahnen Schmerzhemmung RVM Dorsal Horn ACC: anteriorer cingulärer Cortex PAG: periaqueductal gray (5-HT) DLPT: dorsolateral pontine tegmentum (NA) RVM: rostroventral medulla (5-HT) T: Thalamus H: Hypothalamus Schmerz leitendes Neuron Fields HL, et al. Ann Rev Neuro. 1991; 14: Fields HL, et al. Textbook of Pain. 4th ed.1999; 310.
 DLPT: dorsolateral pontine tegmentum (NA) RVM: rostroventral medulla (5-HT) T: Thalamus. H: Hypothalamus. Schmerz leitendes Neuron. Fields HL, et al. Ann Rev Neuro. 1991; 14: Fields HL, et al. Textbook of Pain. 4th ed.1999; 310.")
59
Ursachen peripherer neuropathischer Schmerzen
Verletzung peripherer Nerven durch z. B. mechanische (z. B. Trauma) metabolische/endokrine (z. B. Diabetes, Alkohol, Vit.-B12-Mangel, Hypothyreose) toxische (z. B. Blei, Vincristin) entzündliche/immunologische (z. B. Borreliose) Ursachen Der neuropathische Schmerz kann nach den zugrunde liegenden Ursachen einer möglichen Neuropathie klassifiziert werden. In der Praxis dominieren die Polyneuropathien durch metabolische Ursachen - mit je einem Drittel der Fälle die alkoholische und die diabetische Genese (Brandt 1992). Die klinische Diagnose einer PNP beruht auf der Anamnese- und Beschwerdeschilderung des Patienten und dem klinischen Befund. Nix WA, in Handbuch Chronischer Schmerz, Egle UT et al., Schattauer 2003
 metabolische/endokrine (z. B. Diabetes, Alkohol, Vit.-B12-Mangel, Hypothyreose) toxische (z. B. Blei, Vincristin) entzündliche/immunologische (z. B. Borreliose) Ursachen. Der neuropathische Schmerz kann nach den zugrunde liegenden Ursachen einer möglichen Neuropathie klassifiziert werden. In der Praxis dominieren die Polyneuropathien durch metabolische Ursachen - mit je einem Drittel der Fälle die alkoholische und die diabetische Genese (Brandt 1992). Die klinische Diagnose einer PNP beruht auf der Anamnese- und Beschwerdeschilderung des Patienten und dem klinischen Befund. Nix WA, in Handbuch Chronischer Schmerz, Egle UT et al., Schattauer")
60
Epidemiologie diabetische Polyneuropathie
Die diabetische Neuropathie wird, neben der alkoholtoxischen, als die häufigste Neuropathieform der westlichen Industrieländer angesehen1 Prävalenz diabetische Polyneuropathie: 25 %2 Die diabetische Neuropathie ist eine klinisch-manifeste oder subklinische Erkrankung der peripheren Nerven; sie kann das somatische und/oder autonome Nervensystem betreffen. Es bestehen Assoziationen mit Diabetesdauer, Diabeteseinstellung (Hyperglykämie), diabetischer Retinopathie und Nephropathie, sowie mit arterieller Hypertonie [ / Praxis-Leitlinien / Diabetische Neuropathie / Aktualisierte Version Feb 2005]. Die häufigste Form ist die vorwiegend sensible symmetrische distale Neuropathie. Sie beginnt schleichend und verläuft chronisch progredient. Sie manifestiert sich bevorzugt in den distalen Abschnitten der unteren Extremitäten (längenbezogene Funktionsstörung). Über die Häufigkeit bzw. das Risiko des Auftretens der diabetischen Neuropathie gibt es in der Literatur unterschiedliche Angaben; jeder Diabetiker scheint betroffen zu sein. Auch über die Häufigkeit von Schmerzen im Rahmen einer diabetischen Polyneuropathie gibt es in der Literatur unterschiedliche Angaben. 1. Hopf HC et al, Neurologie in Praxis und Klinik, Thieme Verlag Ziegler D et al, EASD Daousi C. et al, Diabetic Medicine, 21, , 2004
, diabetischer Retinopathie und Nephropathie, sowie mit arterieller Hypertonie [ / Praxis-Leitlinien / Diabetische Neuropathie / Aktualisierte Version Feb 2005]. Die häufigste Form ist die vorwiegend sensible symmetrische distale Neuropathie. Sie beginnt schleichend und verläuft chronisch progredient. Sie manifestiert sich bevorzugt in den distalen Abschnitten der unteren Extremitäten (längenbezogene Funktionsstörung). Über die Häufigkeit bzw. das Risiko des Auftretens der diabetischen Neuropathie gibt es in der Literatur unterschiedliche Angaben; jeder Diabetiker scheint betroffen zu sein. Auch über die Häufigkeit von Schmerzen im Rahmen einer diabetischen Polyneuropathie gibt es in der Literatur unterschiedliche Angaben. 1. Hopf HC et al, Neurologie in Praxis und Klinik, Thieme Verlag Ziegler D et al, EASD Daousi C. et al, Diabetic Medicine, 21, ,")
61
Diabetische Polyneuropathie
Die diabetische Polyneuropathie ist eine klinisch-manifeste oder subklinische Erkrankung der peripheren Nerven, die infolge Diabetes mellitus ohne andere Ursachen auftritt. Es bestehen Assoziationen mit Diabetesdauer, Diabeteseinstellung (Hyperglykämie), diabetischer Retinopathie und Nephropathie, Hypertonie, Hyperlipidämie, Alkohol und Nikotin. Die diabetische Polyneuropathie ist eine klinisch-manifeste oder subklinische Erkrankung der peripheren Nerven, die infolge Diabetes mellitus ohne andere Ursachen auftritt Es bestehen Assoziationen mit Diabetesdauer, Diabeteseinstellung (Hyperglykämie), diabetischer Retinopathie und Nephropathie, Hypertonie, Hyperlipidämie, Alkohol und Nikotin Neuropathie/Aktualisierte Version Feb Reiners K, Haslbeck M, Der Diabetologe 2, 2006, 2:92-103
, diabetischer Retinopathie und Nephropathie, Hypertonie, Hyperlipidämie, Alkohol und Nikotin. Die diabetische Polyneuropathie ist eine klinisch-manifeste oder subklinische Erkrankung der peripheren Nerven, die infolge Diabetes mellitus ohne andere Ursachen auftritt. Es bestehen Assoziationen mit Diabetesdauer, Diabeteseinstellung (Hyperglykämie), diabetischer Retinopathie und Nephropathie, Hypertonie, Hyperlipidämie, Alkohol und Nikotin. Neuropathie/Aktualisierte Version Feb Reiners K, Haslbeck M, Der Diabetologe 2, 2006, 2:")
62
Mehrere Stoffwechselprozesse können zur diabetischen Polyneuropathie beitragen
Diabetes Hyperglykämie Glykolisation Polyolstoffwechsel Oxidativer Stress PKC-b Diabetische Neuropathie Direkte Neurotoxizität Vaskulopathie/Ischämie Modifiziert nach Boulton AJM, et al. Diabetes Care 2004;27:

63
Symptome und Befunde der diabetischen Polyneuropathie
Distal-symmetrische sensomotorische Polyneuropathie ist die häufigste Manifestationsform Symptome Taubheit oder Gefühllosigkeit (eingeschlafen oder das Gefühl eines Sockenbündels unter den Zehen) Kribbeln/Prickeln Dumpfer Schmerz Brennender Schmerz Stechender Schmerz Ungewöhnliche Sensiblität und Empfindlichkeit bei Fußberührungen (Allodynie) Befunde Reduziertes Vibrationsempfinden Abgeschwächter Patellarsehnenreflex und Achillessehnenreflex Reduzierte schützende Empfindungen wie Druck, Hitze und Kälte, Schmerz Verminderte Fähigkeit, die Position von Füßen und Zehen wahrzunehmen Symptome und Anzeichen steigern sich mit der Zeit von distal nach proximal
 Kribbeln/Prickeln. Dumpfer Schmerz. Brennender Schmerz. Stechender Schmerz. Ungewöhnliche Sensiblität und Empfindlichkeit bei Fußberührungen (Allodynie) Befunde. Reduziertes Vibrationsempfinden. Abgeschwächter Patellarsehnenreflex und Achillessehnenreflex. Reduzierte schützende Empfindungen wie Druck, Hitze und Kälte, Schmerz. Verminderte Fähigkeit, die Position von Füßen und Zehen wahrzunehmen. Symptome und Anzeichen steigern sich mit der Zeit von distal nach proximal.")
64
Diagnosekriterien für die sensomotorische Neuropathie bei Diabetes mellitus – NDS
Neuropathie Symptom-Score (NSS) Symptomatik Fuß/Unterschenkel Brennen, Taubheitsgefühl, Parästhesien Schwächegefühl (Ermüdung, Erschöpfung), Krämpfe, Schmerzen Lokalisation Füße oder Unterschenkel oder andere Lokalisation Exazerbation nachts vorhanden oder tagsüber und nachts vorhanden oder nur tagsüber vorhanden Patient wird durch Symptome aus dem Schlaf geweckt Besserung der Symptome beim Gehen oder Stehen oder Sitzen bzw. Hinlegen Gesamtscore: Punkte 2 1 0 Bewertung: 3–4 = leichte Symptome 5–6 = mäßige Symptome 7–10 = schwere neuropathische Symptome Neuropathie/Aktualisierte Version Feb. 2005
 Symptomatik Fuß/Unterschenkel. Brennen, Taubheitsgefühl, Parästhesien. Schwächegefühl (Ermüdung, Erschöpfung), Krämpfe, Schmerzen. Lokalisation. Füße oder. Unterschenkel oder. andere Lokalisation. Exazerbation. nachts vorhanden oder. tagsüber und nachts vorhanden. oder nur tagsüber vorhanden. Patient wird durch Symptome. aus dem Schlaf geweckt. Besserung der Symptome beim. Gehen oder. Stehen oder. Sitzen bzw. Hinlegen. Gesamtscore: Punkte. 2. 1. 0. Bewertung: 3–4 = leichte Symptome. 5–6 = mäßige Symptome. 7–10 = schwere neuropathische Symptome. Neuropathie/Aktualisierte Version Feb")
65
Diagnosekriterien für die sensomotorische Neuropathie bei Diabetes mellitus – NDS
Neuropathie Defizit-Score (NDS) Achillessehnenreflex Reflexe: normal abgeschwächt fehlend Vibrationsempfinden1 Normwerte siehe rechts bei: „Einfache neurologische Untersuchungsmethoden“ Messung am Fußrücken [1. Metatarsale distal] normal abgeschwächt/fehlend Schmerzempfindung Messung am Fußrücken Temperaturempfindung Gesamtscore: Punkte Seite links rechts 0 1 2 Bewertung: 3–5 = leichte neuropathische Defizite 6–8 = mäßige neuropathische Defizite 9–10 = schwere neuropathische Defizite Neuropathie/Aktualisierte Version Feb. 2005
 Achillessehnenreflex. Reflexe: normal. abgeschwächt. fehlend. Vibrationsempfinden1 Normwerte siehe rechts bei: „Einfache neurologische Untersuchungsmethoden Messung am Fußrücken [1. Metatarsale distal] normal. abgeschwächt/fehlend. Schmerzempfindung. Messung am Fußrücken. Temperaturempfindung. Gesamtscore: Punkte. Seite. links rechts. 0 0. 1 1. 2 2. Bewertung: 3–5 = leichte neuropathische Defizite. 6–8 = mäßige neuropathische Defizite. 9–10 = schwere neuropathische Defizite. Neuropathie/Aktualisierte Version Feb")
66
Nicht-Endokrinologen
Die diabetische Polyneuropathie bleibt häufig unentdeckt 10 20 30 40 50 60 70 80 90 100 keine PNP mittelschwere PNP schwere PNP Endokrinologen Nicht-Endokrinologen % der Ärzte, die eine korrekte Diagnose stellen Herman & Kennedy, Diabetes Care 2005; 28:

67
Epidemiologie schmerzhafte diabetische Polyneuropathie
Prävalenz von Schmerzen im Rahmen der diabetischen Polyneuropathie: 16 % 12,5 % der Patienten haben ihrem Arzt nie Schmerzen mitgeteilt 39,3 % wurden nie wegen der Schmerzen behandelt Über die Häufigkeit von Schmerzen im Rahmen einer diabetischen Polyneuropathie gibt es in der Literatur unterschiedliche Angaben. Daousi et al berichteten eine Prävalenz von ca. 16%. Daousi C. et al., Diabetic Medicine, 21, , 2004

68
Klinisches Bild neuropathischer Schmerzen
Parästhesien/Dysästhesien Hyperpathie Brennende Spontanschmerzen einschießende Schmerzattacken Dauerschmerzen evozierte Schmerzen: Hyperalgesie/Allodynie Das klinische Bild neuropathischer Schmerzen ist vielfältig. Es können sowohl Wahrnehmungsdefizite (sensorische Qualitäten fallen aus oder sind abgemildert sind im Sinne einer Hypästhesie oder Hypalgesie) als auch übersteigerte (Schmerz)Wahrnehmungen auftreten. Vom Patienten werden Parästhesien in Form von Ameisenlaufen und Kribbeln beschrieben. Werden sensorische Eindrücke in veränderter unangenehmer Qualität erlebt, sind dies Dysästhesien. Patienten mit neuropathischen Schmerzen berichten nicht selten über ein verzögertes Auftreten der Schmerzempfindung nach Reizung und daran anschließend über eine als übermäßig und stark empfundene Schmerzreaktion, die lange anhält = Hyperpathie. Zum klinischen Bild gehören ebenso plötzlich einschießende Schmerzattacken mit brennendem Charakter, die dann kurz und entweder nur einmalig oder intermittierend auftreten. Statt einem Spontanschmerz kann jedoch auch ein Dauerschmerz vorhanden sein. Als evozierte Schmerzen werden die Hyperalgesie (= ein leicht schmerzhafter Reiz löst deutlich intensivere Schmerzempfindungen aus) und Allodynie * (= ein normalerweise nicht schmerzhafter Reiz löst Schmerz aus) bezeichnet. * Durch die zentrale Sensibilisierung können Impulse aus Aß-Berührungsafferenzen, die beim Gesunden nicht ausreichen, um die WDR-Neurone zu erregen, jetzt eine Erregung und damit eine Schmerzempfindung hervorrufen. Hiemke C, in Handbuch Chronischer Schmerz, Egle UT et al., Schattauer 2003 Tölle TR, Baron R Fortschritte der Medizin 120.Jg.-Originalien Nr. II-III/2002, 49-59
 als auch übersteigerte (Schmerz)Wahrnehmungen auftreten. Vom Patienten werden Parästhesien in Form von Ameisenlaufen und Kribbeln beschrieben. Werden sensorische Eindrücke in veränderter unangenehmer Qualität erlebt, sind dies Dysästhesien. Patienten mit neuropathischen Schmerzen berichten nicht selten über ein verzögertes Auftreten der Schmerzempfindung nach Reizung und daran anschließend über eine als übermäßig und stark empfundene Schmerzreaktion, die lange anhält = Hyperpathie. Zum klinischen Bild gehören ebenso plötzlich einschießende Schmerzattacken mit brennendem Charakter, die dann kurz und entweder nur einmalig oder intermittierend auftreten. Statt einem Spontanschmerz kann jedoch auch ein Dauerschmerz vorhanden sein. Als evozierte Schmerzen werden die Hyperalgesie (= ein leicht schmerzhafter Reiz löst deutlich intensivere Schmerzempfindungen aus) und Allodynie * (= ein normalerweise nicht schmerzhafter Reiz löst Schmerz aus) bezeichnet. * Durch die zentrale Sensibilisierung können Impulse aus Aß-Berührungsafferenzen, die beim Gesunden nicht ausreichen, um die WDR-Neurone zu erregen, jetzt eine Erregung und damit eine Schmerzempfindung hervorrufen. Hiemke C, in Handbuch Chronischer Schmerz, Egle UT et al., Schattauer 2003 Tölle TR, Baron R Fortschritte der Medizin 120.Jg.-Originalien Nr. II-III/2002,")
69
Charakteristika der Schmerzen bei PNP
brennend 80 elektrisch scharf dumpf 64 kribbelnd 60 54 54 50 49 % Patienten 40 20 Um die Schmerzen im Rahmen der diabetischen Polyneuropathie genauer zu untersuchen, wurde eine prospektive Beobachtungsstudie an Patienten mit schmerzhaft diabetischer Polyneuropathie (DPNP) durchgeführt (via Fragebogen). 105 Patienten schickten den Fragebogen ausgefüllt zurück. Die mittlere Schmerzstärke betrug 5.75 (0-10 cm Skala, SD = 2.21). Die Mehrheit der Patienten (53%) berichteten über konstante, tägliche Schmerzen. Die Mehrzahl der übrigen Patienten (30%) klagten ebenfalls über tägliche Schmerzen, die aber intermittierend während des Tages auftreten. 72% der Patienten berichteten, dass sich ihre Schmerzen seit Auftreten verschlechtert haben, bei 12% trat eine Verbesserung ein, bei 15% blieben sie unverändert. Die Qualität der Schmerzen wurde beschrieben als: 64% heiss oder brennend, 54% elektrisch oder schockartig, 54% scharf, 50% dumpf, 49% kribbelnd, 7% kalt, 6% empflindlich, 6% stumpf, 3% „Ameisenlaufen“, 3% pochend, 1% juckend, 1% fest. n = 105 Patienten mit schmerzhafter diabetischer Polyneuropathie (DPNP) Galer et al., Diabetes Research and Clinical Practice 47 (2000) 123–128
 durchgeführt (via Fragebogen). 105 Patienten schickten den Fragebogen ausgefüllt zurück. Die mittlere Schmerzstärke betrug 5.75 (0-10 cm Skala, SD = 2.21). Die Mehrheit der Patienten (53%) berichteten über konstante, tägliche Schmerzen. Die Mehrzahl der übrigen Patienten (30%) klagten ebenfalls über tägliche Schmerzen, die aber intermittierend während des Tages auftreten. 72% der Patienten berichteten, dass sich ihre Schmerzen seit Auftreten verschlechtert haben, bei 12% trat eine Verbesserung ein, bei 15% blieben sie unverändert. Die Qualität der Schmerzen wurde beschrieben als: 64% heiss oder brennend, 54% elektrisch oder schockartig, 54% scharf, 50% dumpf, 49% kribbelnd, 7% kalt, 6% empflindlich, 6% stumpf, 3% „Ameisenlaufen , 3% pochend, 1% juckend, 1% fest. n = 105 Patienten mit schmerzhafter diabetischer Polyneuropathie (DPNP) Galer et al., Diabetes Research and Clinical Practice 47 (2000) 123–128.")
70
Lokalisation der Schmerzen bei DPNP
Fußrücken 54 % Zehen 67 % Hände 39 % Fuß- ballen 69 % Fußsohle 37 % Füße 96 % Um die Schmerzen im Rahmen der diabetischen Polyneuropathie genauer zu untersuchen, wurde eine prospektive Beobachtungsstudie an Patienten mit schmerzhaft diabetischer Polyneuropathie (DPNP) durchgeführt (via Fragebogen). 105 Patienten schickten den Fragebogen ausgefüllt zurück. Die mittlere Schmerzstärke betrug 5.75 (0-10 cm Skala, SD = 2.21). Die Mehrheit der Patienten (53%) berichteten über konstante, tägliche Schmerzen. Die Mehrzahl der übrigen Patienten (30%) klagten ebenfalls über tägliche Schmerzen, die aber intermittierend während des Tages auftreten. 72% der Patienten berichteten, dass sich ihre Schmerzen seit Auftreten verschlechtert haben, bei 12% trat eine Verbesserung ein, bei 15% blieben sie unverändert. -Die Lokalisationen der Schmerzen wurde beschrieben wie folgt: - 96% Füsse: 69% Fussballen, 67% Zehen, 54% Fußrücken, 37% Fußsohle, 37% Wade, 32% Ferse, 25% Knöchel. 39% Hände: 25% Finger, 19% Fingerspitzen, 15% Handfläche, 12% Handrücken, 7% Unterarm, 5% Handgelenk. Ferse 32 % n = 105 Patienten mit schmerzhafter diabetischer Polyneuropathie (DPNP) Galer et al., Diabetes Research and Clinical Practice 47 (2000) 123–128
 durchgeführt (via Fragebogen). 105 Patienten schickten den Fragebogen ausgefüllt zurück. Die mittlere Schmerzstärke betrug 5.75 (0-10 cm Skala, SD = 2.21). Die Mehrheit der Patienten (53%) berichteten über konstante, tägliche Schmerzen. Die Mehrzahl der übrigen Patienten (30%) klagten ebenfalls über tägliche Schmerzen, die aber intermittierend während des Tages auftreten. 72% der Patienten berichteten, dass sich ihre Schmerzen seit Auftreten verschlechtert haben, bei 12% trat eine Verbesserung ein, bei 15% blieben sie unverändert. -Die Lokalisationen der Schmerzen wurde beschrieben wie folgt: - 96% Füsse: 69% Fussballen, 67% Zehen, 54% Fußrücken, 37% Fußsohle, 37% Wade, 32% Ferse, 25% Knöchel. 39% Hände: 25% Finger, 19% Fingerspitzen, 15% Handfläche, 12% Handrücken, 7% Unterarm, 5% Handgelenk. Ferse. 32 % n = 105 Patienten mit schmerzhafter diabetischer Polyneuropathie (DPNP) Galer et al., Diabetes Research and Clinical Practice 47 (2000) 123–128.")
71
Charakteristika der schmerzhaften PNP
Schmerzen und Dysästhesien insbesondere in Ruhe und Nachts (“burning feet” bei Bettwärme) Leitlinie Neuropathischer Schmerz der DGN (Deutsche Gesellschaft Neurologie) 2005, in: Leitlinien für Diagnostik und Therapie in der Neruologie, Hrgs. HC Diener, Aufl.2005
 Leitlinie Neuropathischer Schmerz der DGN (Deutsche Gesellschaft Neurologie) 2005, in: Leitlinien für Diagnostik und Therapie in der Neruologie, Hrgs. HC Diener, Aufl")
72
Freizeit- aktivitäten Modified Brief Pain Inventory (BPI) score 5
Schmerzhafte diabetische Neuropathie senkt die Lebensqualität Lebens- freude Schlaf Mobilität Arbeit Freizeit- aktivitäten Soziale Aktivitäten Allgemeine Aktivität Stimmung Beziehungen Modified Brief Pain Inventory (BPI) score 5 n = 105 mit schmerzhafter DN [% der Personen mit substantieller Beeinträchtigung] % 58 57 56 51 48 43 36 20 40 60 80 100 Galer et al., Diabetes Research and Clinical Practice 47 (2000) 123–128
 score 5. n = 105 mit schmerzhafter DN [% der Personen mit substantieller Beeinträchtigung] % Galer et al., Diabetes Research and Clinical Practice 47 (2000) 123–128.")
73
Therapiealgorithmen bei neuropathischen Schmerzen
Therapie der zugrundeliegenden Ursache Erzielung einer vollständigen oder zumindest teilweisen Analgesie durch pharmakologische und nicht-pharmakologische Methoden Verbesserung der Schmerzbewältigung durch supportive psychologische Therapieverfahren Die Optimierung der Diabeteseinstellung ist der alleinige allgemein akzeptierte Kausalansatz zur Prävention und Therapie der diabetischen Neuropathie. Andere, pathogenetisch begründbare Therapieansätze sind derzeit nicht gesichert. Risikofaktoren müssen erfasst und ggf. behandelt werden. Wichtig ist der Hinweis, dass sich neuropathische Symptome unterschiedlicher Schweregrade innerhalb von Wochen bis Monaten spontan bessern können. Basismassnahmen der Therapie sind: Optimierung der Diabeteseinstellung - Blutdrucknormalisierung (<130/80 mmHg) - Änderungen der Lebensgewohnheiten (Alkohol, Raucher) - Patientenschulung Die Möglichkeiten der symptomatischen medikamentösen Schmerztherapie sind in nachfolgendem Slide aufgeführt. Nicht-medikamentöse Therapie sind z.B. TENS (Transkutane elektrische Nervenstimulation) - SCS (Spinal cord stimulation = neuroelektrische Stimulation des Rückenmarkes) - Physikalische Therapie und Ergotherapie - Psychologische Schmerztherapie. Neuropathie/Aktualisierte Version Feb Tölle TR, Baron R Fortschritte der Medizin 120.Jg.-Originalien Nr. II-III/2002, 49-59
 - Änderungen der Lebensgewohnheiten (Alkohol, Raucher) - Patientenschulung. Die Möglichkeiten der symptomatischen medikamentösen Schmerztherapie sind in nachfolgendem Slide aufgeführt. Nicht-medikamentöse Therapie sind z.B. TENS (Transkutane elektrische Nervenstimulation) - SCS (Spinal cord stimulation = neuroelektrische Stimulation des Rückenmarkes) - Physikalische Therapie und Ergotherapie - Psychologische Schmerztherapie. Neuropathie/Aktualisierte Version Feb Tölle TR, Baron R Fortschritte der Medizin 120.Jg.-Originalien Nr. II-III/2002,")
74
Erfassung der Schmerzintensität
VAS = Visuelle Analogskala NRS = Numerische Ratingskala (Likert Skala) Vor der Schmerztherapie muss eine sorgfältige Schmerzanamnese erhoben werden und eine Schmerzmessung erfolgen. Bewährte und einfach anzuwendende Verfahren zur Einschätzung der Schmerzintensität sind die Visuelle Analogskala (VAS) und numerische Ratingskalen (NRS). Auf einer Linie von 10 cm sind die Endpunkte mit ‚kein Schmerz‘ und ‚unerträgliche Schmerzen‘ bezeichnet. Der Patient markiert die Stelle oder die Zahl, die seiner subjektiv empfundenen Schmerzstärke entspricht. Für den Patienten bedeutet eine Reduktion der Schmerzen um 30% eine klinisch relevante Verbesserung; eine Reduktion um 50% korreliert mit der Patienteneinschätzung „es geht mir sehr viel besser“. Bei chronischen Schmerzen bedeutet eine Reduktion der Schmerzen um 30 % eine klinisch relevante Verbesserung; eine Reduktion um 50 % korreliert mit der Patienteneinschätzung „sehr viel besser“.1 1) Rowbotham MC, 2001, Pain, 94:
 Vor der Schmerztherapie muss eine sorgfältige Schmerzanamnese erhoben werden und eine Schmerzmessung erfolgen. Bewährte und einfach anzuwendende Verfahren zur Einschätzung der Schmerzintensität sind die Visuelle Analogskala (VAS) und numerische Ratingskalen (NRS). Auf einer Linie von 10 cm sind die Endpunkte mit ‚kein Schmerz‘ und ‚unerträgliche Schmerzen‘ bezeichnet. Der Patient markiert die Stelle oder die Zahl, die seiner subjektiv empfundenen Schmerzstärke entspricht. Für den Patienten bedeutet eine Reduktion der Schmerzen um 30% eine klinisch relevante Verbesserung; eine Reduktion um 50% korreliert mit der Patienteneinschätzung „es geht mir sehr viel besser . Bei chronischen Schmerzen bedeutet eine Reduktion der Schmerzen um 30 % eine klinisch relevante Verbesserung; eine Reduktion um 50 % korreliert mit der Patienteneinschätzung „sehr viel besser .1. 1) Rowbotham MC, 2001, Pain, 94:")
75
Nicht alle Wirkstoffe sind zur Schmerzbehandlung zugelassen!
Symptomatische, pharmakologische Behandlungsmöglichkeiten (1) Antidepressiva (z. B. Trizyklika, SNRIs*, SSRIs*, SSNRIs*) Antikonvulsiva (z. B. Gabapentin, Pregabalin, Carbamazepin, Oxcarbazepin, Lamotrigin) Opioide (Tramadol, Oxycodon) -Liponsäure lokale Therapeutika (Capsaicin) Nicht alle Wirkstoffe sind zur Schmerzbehandlung zugelassen! Die Kenntnis der Schmerzstoffe und –mediatoren und deren funktionelles Zusammenwirken zeigt vielfältige pharmakotherapeutische Zugänge für die Behandlung von neuropathischen Schmerzen auf. Manipulationen mit unterschiedlichstem pharmakologischen Wirkprinzip haben – auch beim neuropatischen Schmerz – Eingang in die Klinik gefunden. Die Effektivität der symptomatischen, pharmakologischen Behandlungsmöglichkeiten, die im Rahmen der evidenzbasierten, plazebokontrollierten, doppelblinden Untersuchungen abgeleitet werden kann, gibt erste Orientierung für den Einsatz verschiedener Substanzgruppen. Auf der Grundlage der verfügbaren kontrollierten Studien kann eine pharmakologische Basistherapie neuropathischer Schmerzsymptome mit den o.g. Medikamenten empfohlen werden. Nach klinischer Erfahrung kann auch eine Kombination aus zwei oder drei Wirkstoffen sinnvoll sein. Mit einer medikamentösen Therapie ist eine 50-80%ige Schmerzreduktion zu erwarten, eine Schmerzfreiheit kann fast nie erreicht werden. Bei allen medikamentösen Optionen sprechen ca % der Patienten nur unzureichend auf die Therapie an (< 50%ige Schmerzreduktion) oder leiden an nicht tolerierbaren Nebenwirkungen [Leitlinie Neuropathischer Schmerz der DGN (Deutsche Gesellschaft Neurologie) 2005 (in press)]. . * SNRI = selektiver Noradrenalin- (NA) Wiederaufnahmehemmer * SSRI = selektiver Serotonin-Wiederaufnahmehemmer * SSNRI = selektiver Serotonin- und NA-Wiederaufnahmehemmer, z. B. Duloxetin Nach Leitlinie Neuropathischer Schmerz der DGN (Deutsche Gesellschaft Neurologie) 2005 (in press) Nach Sarholz M, Assion HJ, Psychopharmakotherapie 2005; 12:77-82
 Antidepressiva (z. B. Trizyklika, SNRIs*, SSRIs*, SSNRIs*) Antikonvulsiva (z. B. Gabapentin, Pregabalin, Carbamazepin, Oxcarbazepin, Lamotrigin) Opioide (Tramadol, Oxycodon) -Liponsäure. lokale Therapeutika (Capsaicin) Nicht alle Wirkstoffe sind zur. Schmerzbehandlung zugelassen! Die Kenntnis der Schmerzstoffe und –mediatoren und deren funktionelles Zusammenwirken zeigt vielfältige pharmakotherapeutische Zugänge für die Behandlung von neuropathischen Schmerzen auf. Manipulationen mit unterschiedlichstem pharmakologischen Wirkprinzip haben – auch beim neuropatischen Schmerz – Eingang in die Klinik gefunden. Die Effektivität der symptomatischen, pharmakologischen Behandlungsmöglichkeiten, die im Rahmen der evidenzbasierten, plazebokontrollierten, doppelblinden Untersuchungen abgeleitet werden kann, gibt erste Orientierung für den Einsatz verschiedener Substanzgruppen. Auf der Grundlage der verfügbaren kontrollierten Studien kann eine pharmakologische Basistherapie neuropathischer Schmerzsymptome mit den o.g. Medikamenten empfohlen werden. Nach klinischer Erfahrung kann auch eine Kombination aus zwei oder drei Wirkstoffen sinnvoll sein. Mit einer medikamentösen Therapie ist eine 50-80%ige Schmerzreduktion zu erwarten, eine Schmerzfreiheit kann fast nie erreicht werden. Bei allen medikamentösen Optionen sprechen ca % der Patienten nur unzureichend auf die Therapie an (< 50%ige Schmerzreduktion) oder leiden an nicht tolerierbaren Nebenwirkungen [Leitlinie Neuropathischer Schmerz der DGN (Deutsche Gesellschaft Neurologie) 2005 (in press)]. . * SNRI = selektiver Noradrenalin- (NA) Wiederaufnahmehemmer * SSRI = selektiver Serotonin-Wiederaufnahmehemmer. * SSNRI = selektiver Serotonin- und NA-Wiederaufnahmehemmer, z. B. Duloxetin. Nach Leitlinie Neuropathischer Schmerz der DGN (Deutsche Gesellschaft Neurologie) 2005 (in press) Nach Sarholz M, Assion HJ, Psychopharmakotherapie 2005; 12:")
76
Diabetische Neuropathie
Symptomatische, pharmakologische Behandlungsmöglichkeiten (2) Symptomatische Schmerztherapie Medikamentöse Behandlung1 Antidepressiva z. B. Amitriptylin, Clomipramin, Imipramin (TCA), Duloxetin (SSNRI) Antikonvulsiva z. B. Carbamazepin2, Gabapentin2, Pregabalin Antioxidantien Alpha-Liponsäure, intravenös Opiode z. B. Tramadol, Oxycodon Praxis-Leitlinien Diabetische Neuropathie In den Praxisleitlinien ‚Diabetische Neuropathie‘ der DDG (Deutsche Diabetes Gesellschaft) werden zur symptomatischen Schmerztherapie die o.g. Medikamente empfohlen. Alle genannten Pharmaka sind in Deutschland zugelassen. 1 Es werden nur in Deutschland zugelassene Pharmaka angegeben 2 Einschleichende Dosierung beachten Neuropathie/Aktualisierte Version Feb. 2005
 Symptomatische Schmerztherapie. Medikamentöse Behandlung1. Antidepressiva z. B. Amitriptylin, Clomipramin, Imipramin (TCA), Duloxetin (SSNRI) Antikonvulsiva z. B. Carbamazepin2, Gabapentin2, Pregabalin. Antioxidantien Alpha-Liponsäure, intravenös. Opiode z. B. Tramadol, Oxycodon. Praxis-Leitlinien. Diabetische Neuropathie. In den Praxisleitlinien ‚Diabetische Neuropathie‘ der DDG (Deutsche Diabetes Gesellschaft) werden zur symptomatischen Schmerztherapie die o.g. Medikamente empfohlen. Alle genannten Pharmaka sind in Deutschland zugelassen. 1 Es werden nur in Deutschland zugelassene Pharmaka angegeben. 2 Einschleichende Dosierung beachten. Neuropathie/Aktualisierte Version Feb")
77
Krankheitsbild – Zusammenfassung
Source: Krankheitsbild – Zusammenfassung Diabetes-Patienten leiden langfristig an zahlreichen Folgeerkrankungen, darunter an sensiblen Polyneuropathien Schmerzempfindungen unterschiedlichster Art, Ausprägung und Dauer treten im Rahmen diabetischer Polyneuropathien häufig auf Pharmakotherapeutisch sind zahlreiche Angriffspunkte möglich; auch Antidepressiva sind für die Behandlung von Bedeutung Review: Reviewer Memo: Slide Modified: on: 9/7/2004 2:19:56 PM SL1 Rev: 528 Memo: RJS

78
...Blutzuckersenkende Medikamente
Insulinsensitizer vom „ Gliazontyp „ als Ausblick : Regulation über den PPARγ-Rezeptor PPARγ Muskel Glukoseverwertung PPARγ Fettgewebe Fettspeicherung Reduzierte Lipolyse Freie Fettsäuren Thiazolodin- dione Euglykämie Hepatische Glukoseaufnahme Hepatischer Glukoseausstoss PPARγ Leber

86
Dr. Armin Sammler, Diabetologe DDG

87
Vertreter der Glitazone
Avandia (Rosiglitazone) Actos (Pioglitazone) Dr. Armin Sammler, Diabetologe DDG
 Actos (Pioglitazone) Dr. Armin Sammler, Diabetologe DDG.")
93
körperliche Aktivität
Alle genannten Medikamente können nach Nutzen-Risiko-Abwägung auch mit Insulin kombiniert werden Voraussetzung einer guten, normnahen Stoffwechselführung ( Glukose / Fett ) bleibt die adäquate Ernährung und körperliche Aktivität
 bleibt die adäquate Ernährung. und. körperliche Aktivität.")
94
Ohne Einhaltung bestimmter Ernährungsregeln auch unter innovativen Medikamenten kein Erreichen der Behandlungsziele! Behandlungsziele: Blutzuckertagesprofile und HbA1c im definierten Bereich

95
Schulung mit Schwerpunkt Ernährung als Hauptsäule der Behandlung des Diabetes

96
Das Endocannabinoid-System
1. Was sind Endocannabinoide und Cannabinoid-Rezeptoren? 2. Rolle des Endocannabinoid-Systems in der Kontrolle der Energiebilanz 3. Blockade des Cannabinoid-Rezeptors CB1 bei kardiometabolischem Risiko

97
1. Was sind Endocannabinoide und Cannabinoid-Rezeptoren?

98
Eigenschaften der Endocannabinoide
sind fettlöslich werden « bei Bedarf » synthetisiert wirken lokal (parakrin und/oder autokrin) werden schnell wieder abgebaut wirken als retrograde Neurotransmitter im Nervensystem
 werden schnell wieder abgebaut. wirken als retrograde Neurotransmitter im Nervensystem.")
99
2. Rolle des EC-Systems in der Kontrolle der Energiebilanz

100
CB1-Blockade verringert Verlangen nach Genussnahrung
Sucrose Stückchen (/Ratte) 1,0 ± 0.4** 1,6 ± 0.2** 2,1 ± 0.2 3,1 ± 0.4 3,0 mg/kg 1,0 mg/kg 0,3 mg/kg Kontrolle Rimonabant Arnone M et al, Psychopharmacology 1997;132:104–106 Rimonabant und die Aufnahme von Sucrose **p<0,01 CB1-Blockade verringert Verlangen nach Genussnahrung
 1,0 ± 0.4** 1,6 ± 0.2** 2,1 ± ,1 ± ,0 mg/kg. 1,0 mg/kg. 0,3 mg/kg. Kontrolle. Rimonabant. Arnone M et al, Psychopharmacology 1997;132:104–106. Rimonabant und die Aufnahme von Sucrose. **p<0,01. CB1-Blockade verringert Verlangen nach Genussnahrung.")
101
Das EC-System und der Hypothalamus

102
CB1-Blockade als vielverspechender therapeutischer Ansatz
Beobachtungen: CB1-Aktivierung induziert Hunger und Essen 2. EC-System ist überaktiviert bei Fettleibigkeit CB1-Blockade als vielverspechender therapeutischer Ansatz Ergo:

103
CB1-Blockade beeinflusst metabolische Prozesse in peripheren Geweben !

104
Das EC-System und die Adipozyten

105
CB1-Blocker Rimonabant stimuliert die Adiponektin-Produktion in Adipozyten
Bensaid M et al. Mol Pharm 2003;63:908–914 Adiponektin: Fettgewebe-spezifisches, im Blut zirkulierendes Protein (= Adipokin) Zentraler Regulator des Fett- und Zuckerstoffwechsels Erhöhte Adiponektin-Mengen: bessere Insulinsensitivität, hemmt Atherogenese
 Zentraler Regulator des Fett- und Zuckerstoffwechsels. Erhöhte Adiponektin-Mengen: bessere Insulinsensitivität, hemmt Atherogenese.")
106
3. Blockade des CB1-Rezeptors bei kardiometablischem Risiko

107
Überaktivität des EC-Systems Erhöhte Nahrungszufuhr
Effekte einer Überaktivität des EC-Systems Übermaß an Nahrung → Fettleibigkeit Überaktivität des EC-Systems Multiple Wirkorte Adipozyten, Leber, GI-Trakt Fettakkumulation, Hunger Nucleus accumbens Motivation zu essen Hypothalamus Appetit Insulinresistanz Ruheglukose Adiponektin HDL-C Triglyceride Erhöhte Nahrungszufuhr

108
Multiple Wirkorte und Effekte der CB1-Blockade
Mechanismen Behandelt Nucleus Accumbens Hypothalamus Essen Körpergewicht Intra-abdominale Adipositas Adipozyten Futile cycles Lipolyse Adiponektin Körpergewicht Dyslipidemie Insulinresistanz Leber Lipogenese Dyslipidemie Insulinresistenz Gastrointestinal-Trakt Sättigungssignale Körpergewicht Intra-abdominale Adipositas Muskeln Glucoseaufnahme Energieverbrauch Insulinresistenz Di Marzo et al 2001; Ravinet Trillou et al 2003; Cota et al 2003; Pagotto et al 2005; Van Gaal et al 2005; Liu et al 2005; Osei-Hyiaman et al 2005; Jbilo et al 2005

109
Ich bedanke mich für Ihre Aufmerksamkeit.
Unter steht der Vortrag zum Download bereit

Ähnliche Präsentationen
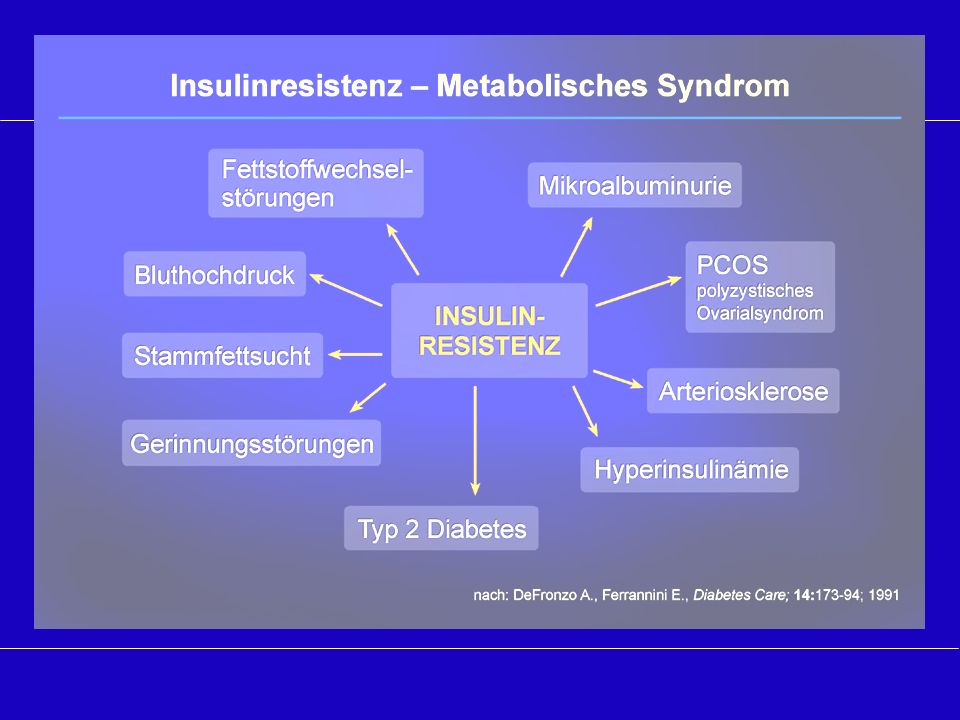
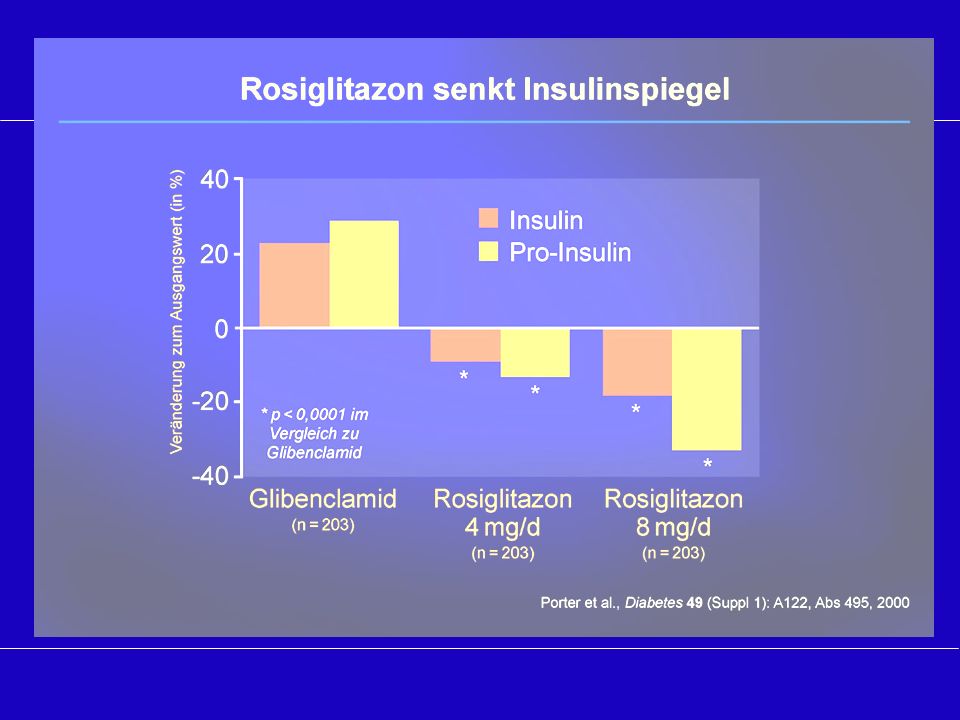



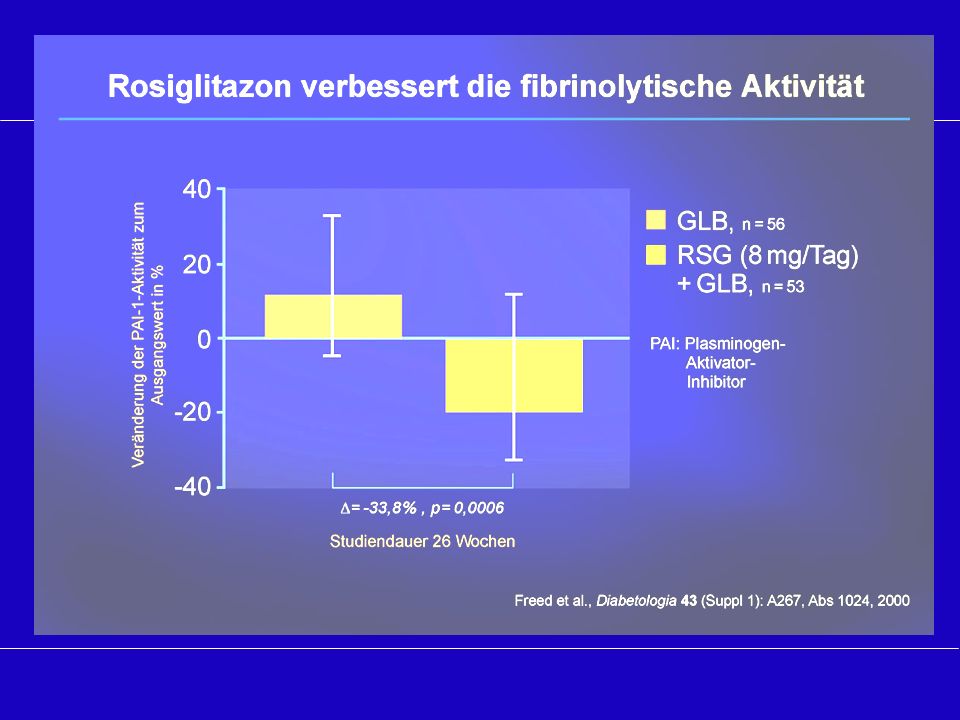
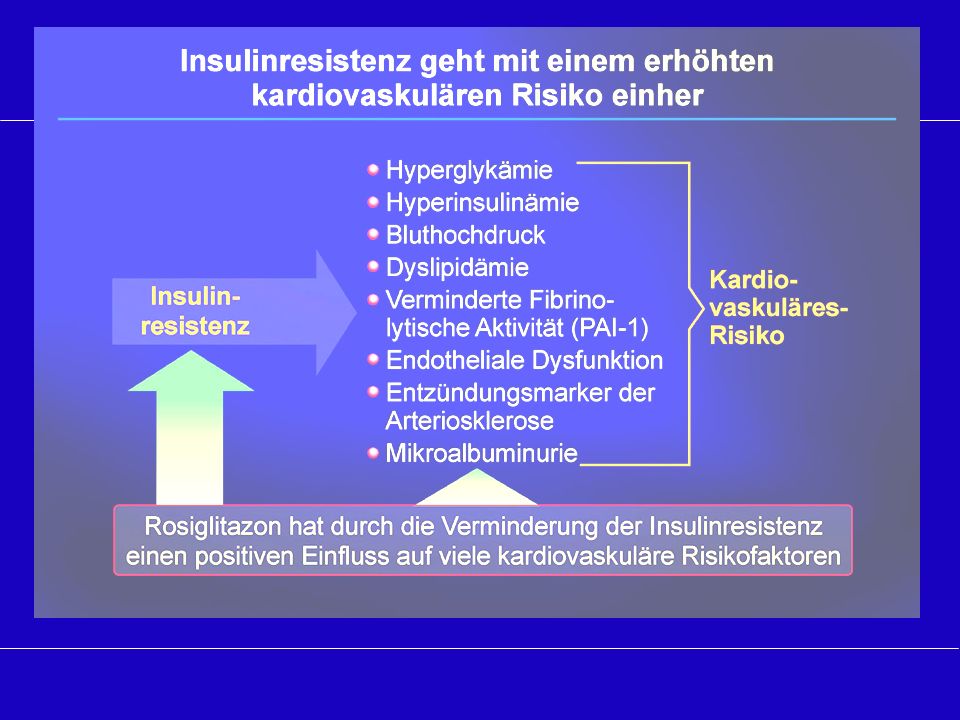

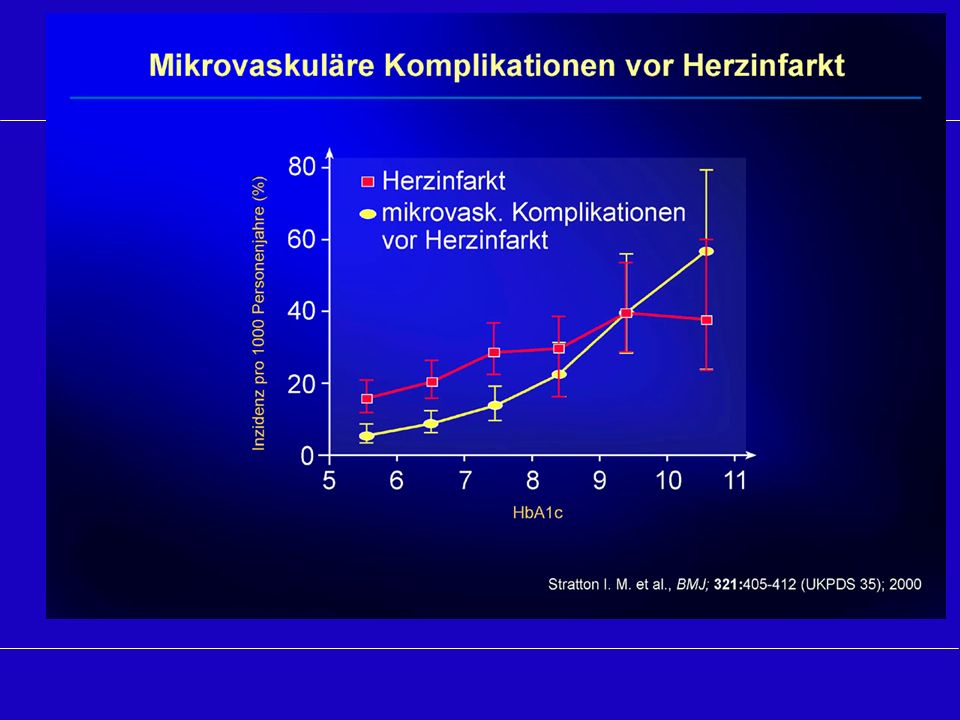
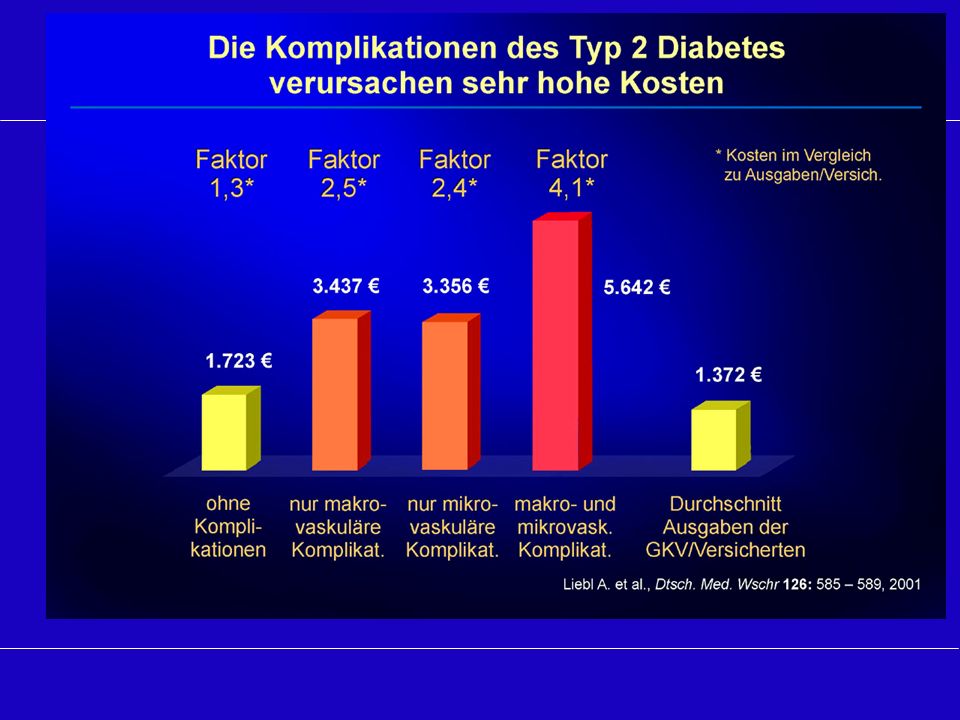
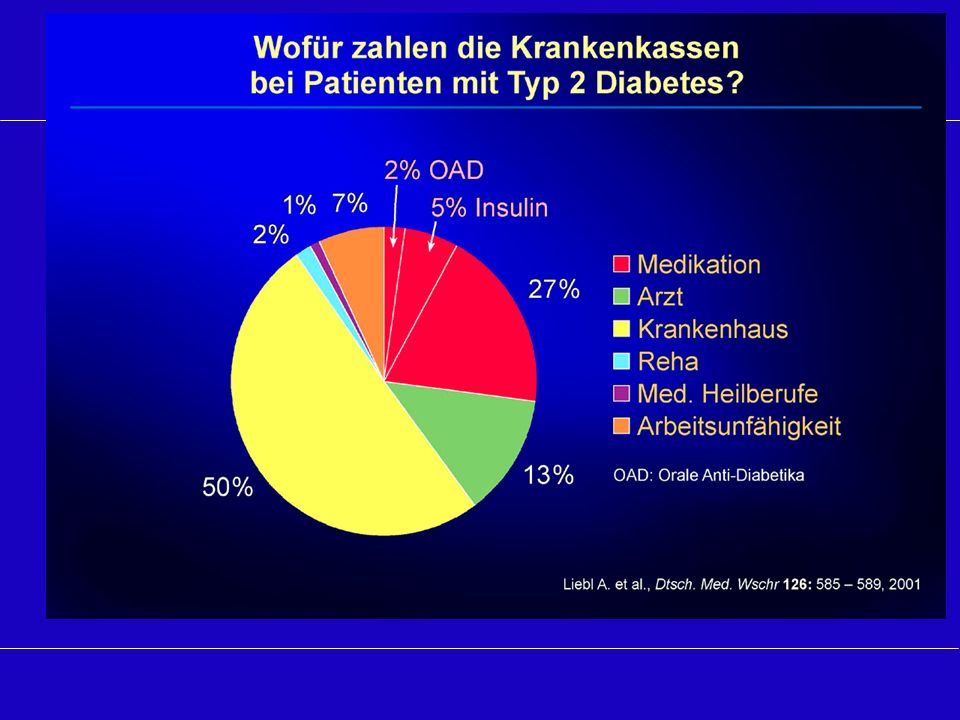




 bei der>")



 BENCHMRK: Blocking integrase in treatment Experienced patients.>")











 bei Herzinsuffizienz>")