Präsentation herunterladen
Die Präsentation wird geladen. Bitte warten

1
Mikrobielle Modelle in der Neuropathologie

2
Warum?

3
"Alle wollen es werden, keiner will es sein: alt"
"Alle wollen es werden, keiner will es sein: alt". Denn Langlebigkeit hat ihren Preis. Bis zum Jahre 2050 wird jeder Dritte in Deutschland über 60 Jahre alt sein. Die Zahl der altersbedingten Krankheiten, besonders der Demenzkrankheiten, für die es bisher keine wirksame Therapie oder Heilung gibt, wird stark ansteigen.

4
Demographie Die bekannteste und mit etwa 60 Prozent weitaus häufigste Ursache einer Demenz ist die Alzheimer-Krankheit. An zweiter Stelle stehen mit einem Anteil von knapp 20 Prozent die vaskulären Demenzen, gefolgt von einer weiteren degenerativen Gehirnerkrankung, der Parkinson-Krankheit. Für die übrigen europäischen Länder, Nordamerika und Japan ergeben sich Gesamtzahlen für Demenzkrankheiten, die mindestens zehnmal so hoch wie in Deutschland sind. Für die USA allein werden derzeit vier Millionen Alzheimer-Erkrankte angenommen; bis zum Jahr 2050 wird dort mit einer Zunahme auf 14 Millionen gerechnet. Seltenere Erkrankungen: Huntington, frontemporale Demenz, ALS, Friedreichs Ataxie, Prione, Spartin/Spastin

5
Prionenerkrankungen (Rinderwahn, Creutzfeld-Jakob)
Infektiöses Protein: Eine abnormale Konformation macht das humane PRP Prion Proteaseresistent und initiiert seine Aggregation. Dieses abnormale Prion kann normale Proteine rekrutieren, die Aggregate können wachsen und andere Zellen infizieren. Hefe hat Rinderwahnsinn: Proteine mit Prionendomäne. Mit extrem geriner Wahrscheinlichkeit kann sich so ein Protein falsch falten und den Keim für Aggregate bilden und dann weitere Proteine rekrutieren. Diese werden an Tochterzellen weitergegeben und infizieren diese in einer nicht-mendelschen Form der Vererbung.

6
Prionenerkrankungen (Rinderwahn, Creutzfeld-Jakob)
Humanes PRP kann sich in Hefe in die Protease-Resistente Form umwandeln und aggregiert. Weitere Ergebnisse aus Hefe: Normalerweise wird PRP cotranslationell ins ER geschleust und dann zur Plasmamembran geschickt. Ein kleiner Teil wird sekundär ins Cytosol geschleust wo er sofort vom Proteasom degradiert wird. Eine kleine Fraktion entkommt dem Proteasom und faltet sich zum Todesprion. Der Mechansimus konnte also in Hefe aufgeklärt werden und wurde dann im Mausmodell bestätigt.

7
Parkinson`s disease (PD)
Ist definiert durch Verlust von dopaminergen Neuronen in der Substantia Nigra, und cytoplasmatischen Inklusionen aus Alpha Synuclein (Lewy bodies). Zur Zeit hocherfolgreiches Hefemodell: Zwei Punktmutationen in alpha Synuclein bewirken vererbbaren frühen Ausbruch der Krankheit. In Hefe bilden sich auch dort Protofibrillen nach Expression mutierter Formen des humanen alpha Synuclein aus. (Nicht aber bei der Expression von Wildtypformen). Expressionsniveau und Art der Mutation korreliert mit Tod.
. Zur Zeit hocherfolgreiches Hefemodell: Zwei Punktmutationen in alpha Synuclein bewirken vererbbaren frühen Ausbruch der Krankheit. In Hefe bilden sich auch dort Protofibrillen nach Expression mutierter Formen des humanen alpha Synuclein aus. (Nicht aber bei der Expression von Wildtypformen). Expressionsniveau und Art der Mutation korreliert mit Tod.")
8
Alzheimer Kennzeichen: Calcium im Cytosol, Tao Hyperphosphorylierung, ß-Amyloid-Plaques, ROS, mitochondriale Dysfunktion und Zelltod. Könnte auf mitochondriale Fragmentierung zurückzuführen sein, die gerade in Hefe studiert wird. Hefemodell im Aufbau. Intra oder extrazellulär toxisch? Detoxifizierend oder toxische Agreggate?

9
Polyglutamin-induzierte Krankheiten (Chorea Huntington).
Mutationen in Hefegenen, die für DNA Reparatur zuständig sind, erhöhen die genetische Instabilität in Bereichen, die für die Bildung von Polyglutaminen verantwortlich sind. (Trinucleotid-wiederholungen, in diesem fall CAG). In Neuronen, die unter PolyQ zugrunde gehen, gibt es eine starke Ausdehnung von CAG Wiederholungen im Genom. Expression von PolyQ in Hefe zeigte: Längen, die im Säuger Krankheit auslösen, sind in Hefe unlöslich! HSP70 und HSP40 Überexpression verhinderte die Aggregation in vivo und vitro.
. In Neuronen, die unter PolyQ zugrunde gehen, gibt es eine starke Ausdehnung von CAG Wiederholungen im Genom. Expression von PolyQ in Hefe zeigte: Längen, die im Säuger Krankheit auslösen, sind in Hefe unlöslich! HSP70 und HSP40 Überexpression verhinderte die Aggregation in vivo und vitro.")
10
Polyglutamin-induzierte Krankheiten.
In Hefe wurde gezeigt, dass es zwei distinkte Prozesse gibt: Nukleierung und Wachstum von PolyQ-Aggregaten: Defekte im Wachstum führte zur Initiierung vieler Aggregate. Defekte in der Initiiierung zum schnellen Wachstum weniger Aggregate. Welcher Prozess ist toxisch und welcher detoxifizierend? Sowohl zuviel als auch zuwenig HSP70 und HSP104 inhibiert die Bildung der Aggregate und das Wachstum. Nukleierung erfordert ein neues Prion: RNQ1

11
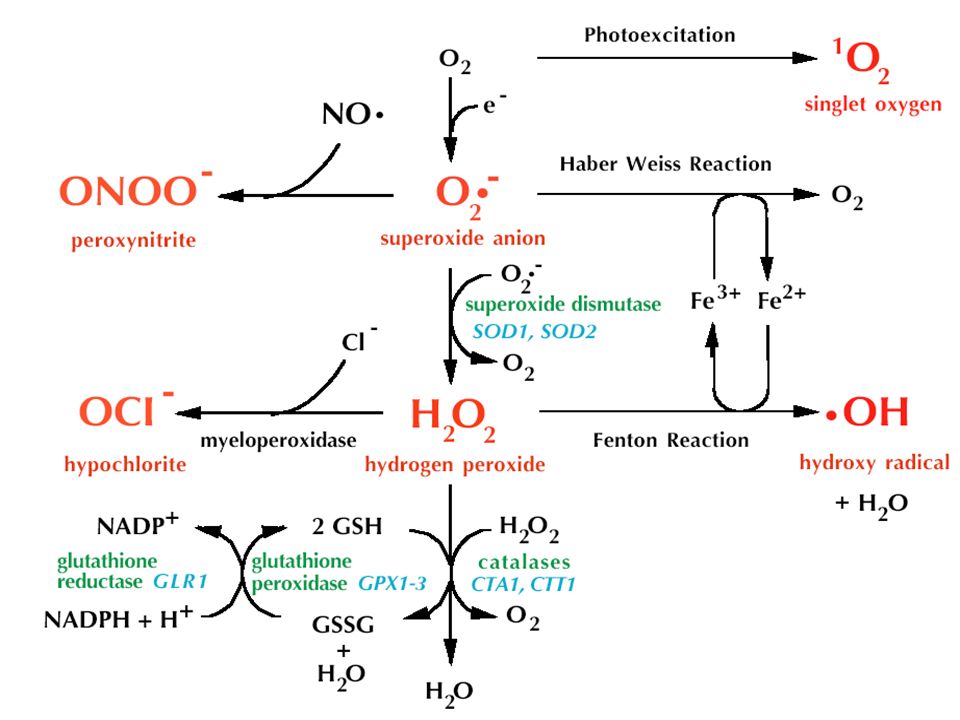
Sauerstoffstress in neurodegenerativen Erkrankungen: Ursache oder Folge?
Post-Mortem Gehirnuntersuchungen an Patienten mit Alzheimer, Parkinson, Amyothropischer Lateralsklerorse (ALS) zeigen deutliche Marker von oxidativem Stress: Nämlich durch Lipidoxidation verursacht:4-Hydroxynonenal, Malondialdehyd oder durch Proteinoxidation verursacht, nämlich Nitrierung von Proteinen. Der Sauersrstoffstress ist zielspezifisch: Nitrierung von Tyrosin in alpha-synuclein der Lewy bodies, die man in PD beobachtet oder von Tyrosinen des Tao Proteins in AD.
 zeigen deutliche Marker von oxidativem Stress: Nämlich durch Lipidoxidation verursacht:4-Hydroxynonenal, Malondialdehyd oder durch Proteinoxidation verursacht, nämlich Nitrierung von Proteinen. Der Sauersrstoffstress ist zielspezifisch: Nitrierung von Tyrosin in alpha-synuclein der Lewy bodies, die man in PD beobachtet oder von Tyrosinen des Tao Proteins in AD.")
12
Sauerstoffstress in neurodegenerativen Erkrankungen: Ursache oder Folge?
Wegen seines hohen metabolischen Umsatz und der relativ geringen Reparaturkapazizät könnte das Gehirn für Ros besonders anfällig sein. Die Antioxidams enzyme SOD, Katalase, Gluthation Peroxidase, und Gluthation Reduktase zeigen erniedrigte Aktivitäten im Gehirn von Patienten mit neurodegenerativen Erkrankungen

13
Parkinson und ROS In PD ist GSH-Depletion der früheste Marker und geht Hand in Hand mit der Degeneration der Substantia nigra. Stärke der Depletion korreliert mit Degeneration. Eisenkonzentrationen sind ebenfalls erhöht (potenzieren Sauerstoffstress durch Fenton-Reaktion) Chelattherapie? SOD1 sowie SOD2 Knock out Mäuse sind empfindlich gegenüber der PD auslösenden Substanz MPTP, wohingegen Transgene mit Mutationen in SOD1 sowie SOD2 vor dieser Substanz schützen
 Chelattherapie SOD1 sowie SOD2 Knock out Mäuse sind empfindlich gegenüber der PD auslösenden Substanz MPTP, wohingegen Transgene mit Mutationen in SOD1 sowie SOD2 vor dieser Substanz schützen.")
15
ALS, Alzheimer und ROS Transgene Mäuse, die mutierte Form von SOD1 exprimieren (Human ALS), zeigen nicht nur Marker von Sauerstoffstress, sondern auch Motoneurodegeneration. Metalchelatoren verhindern AD in Mäusen, die das Amyloid-precursor Protein exprimieren Fazit: Antioxidans-Therapien für Patienten. Vitamin E hilft bei AD und ALS
, zeigen nicht nur Marker von Sauerstoffstress, sondern auch Motoneurodegeneration. Metalchelatoren verhindern AD in Mäusen, die das Amyloid-precursor Protein exprimieren. Fazit: Antioxidans-Therapien für Patienten. Vitamin E hilft bei AD und ALS.")
16
Alter und Neurodegeneration
ROS induzierte Veränderungen von Proteinen wie alpha-synuclein in PD, beta-Amyloid in AD und SOD1 in ALS führen vermutlich zur Überlastung des Proteasoms, was schließlich zur Aggregation dieser Proteine führt.Autophagie? Warum sind neurodegenerative Erkrankungen Alterserkrankungen? Im Alter nimmt der Oxidative Stress im Gehirn zu. HSPs, die vor ROS schützen, nehmen ab, die Anzahl der Proteine, die vom Proteasom nicht mehr abgebaut werden kann, steigt. Die Kapazität der Autophagie sinkt. Induktion von HSPs verbessert den Krankheitsverlauf von ALS Mäusen und erhöhte HSP70 Expression schützt vor PD

17
Apoptose und Neurodegeneration
Rolle von PCD nicht klar, obwohl es im Falle von AD an Beta-amyloid induzierter mitochondrialer Fehlfunktion liegen kann, dass die Zellen zugrunde gehen (ähnliches gilt für PD). Diese erhöht Suszeptibilität für Oxstress könnte Zelltod induzierten. Außerdem wurde In ALS-Mäusen NO-abhängige Caspaseaktivierung festgestellt. MPTP führt zu DNA Schäden, P53 aktivierung und dann Bax Aktivierung. P53 knock out mach die Mäuse resistenter gegen MPTP. GSH Depletion führt zu PCD in dopaminergen Neuronen. Erhöhung des GSH levels vermindert dopaminerge Neurotoxizität.
. Diese erhöht Suszeptibilität für Oxstress könnte Zelltod induzierten. Außerdem wurde In ALS-Mäusen NO-abhängige Caspaseaktivierung festgestellt. MPTP führt zu DNA Schäden, P53 aktivierung und dann Bax Aktivierung. P53 knock out mach die Mäuse resistenter gegen MPTP. GSH Depletion führt zu PCD in dopaminergen Neuronen. Erhöhung des GSH levels vermindert dopaminerge Neurotoxizität.")
18
ROS, Mitochondrien und Neurodegeneration
Herbizide wie Rotenon können PD auslösen. MPTP und Rotenon inhibieren Komplex I der Atmungskette, was zu Generierung von Superoxid sowie zu verminderter mitochondrialer Funktion und somit vermindertem ATP führt. ATP braucht man aber, um Dopamin in dopaminerge Vesikel zu verpacken, was also zu PD führt. Dopamin-Oxidation selbst führt wiederum zu Komplex I Hemmung. Rotenon führt auch zur Bildung von alpha Synuclein und Lewy bodies Transgene ALS Mäuse zeigen reduzierte mitochondriale Kapazität und vermehrte ROS Schäden an Mitos. Fazit: Cycle of death zwische ROS und Neurodegeneration, deshalb helfen Antioxidantien.

19
Proteinfaltung und Neurodegeneration
Generell: Aggregation von falsch gefalteten Proteinen löst neurodegenerative Krankheiten aus. Dabei bilden sich unlösliche Inclusion-bodies aus. Ibs entstehen, weil denaturierte Proteine gerne miteinander aggregieren, trotzdem: Ein Prozess mit vielen Schritten. Meistens am Centrosom oder im Kern. Vermutlich wird Proteinschrott über microtubuli-abhängigen Transport in Richtung Centrosom gelenkt. Es gibt drei Möglichkeiten, falsch gefaltete Proteine zu entsorgen: Autophagie, HSPs und das Proteasom. Beide letzteren aggregieren in Ibs. Störungen in beiden Bereichen spielen eine wichtige rolle fürs Alter und Neurodegeneration. Chaperone und Proteasom sind evolutionär extrem gut konserviert. Säuger und Hefezellen antworten auf Proteinaggregaten mit schneller Induktion des Proteasoms und Induktion von HSPs

20
Sind Ibs die Ursache der Neurodegeneration?
Es gibt auch Leute, die behaupten, dass Ibs ein Abwehrmechanismus sind um das Cytosol vor falsch gefalteten Proteinen zu reinigen. PolyQ aggregation gibt es auch in Hefe (Huntingtons disease). Dafür ist ein intaktes Microtubulisystem nötig. Apoptose und Senesenz sind wichtige Auslöser/Effektoren von Neurodegeneration. Beides ist in Hefe vorhanden. Jahrelange Arbeit haben eine handvoll Gene ergeben, die Proteinaggregation induzierte Neurotoxizität regulieren (C.elegans, Drosophlila). In Hefe könnte man in Monaten hunderte Gene finde. Trotzdem sollten diese dann in höheren Modellen mit echten Gehirnen (Drosophila) verifiziert werden. Schliesslich an intellektuellen Mausmodellen.
. Dafür ist ein intaktes Microtubulisystem nötig. Apoptose und Senesenz sind wichtige Auslöser/Effektoren von Neurodegeneration. Beides ist in Hefe vorhanden. Jahrelange Arbeit haben eine handvoll Gene ergeben, die Proteinaggregation induzierte Neurotoxizität regulieren (C.elegans, Drosophlila). In Hefe könnte man in Monaten hunderte Gene finde. Trotzdem sollten diese dann in höheren Modellen mit echten Gehirnen (Drosophila) verifiziert werden. Schliesslich an intellektuellen Mausmodellen.")
21
Friedreich`s Ataxia (FRDA)
Autosomal-rezessive neurodegenerative Krankheit. Verlängertes Trinucleotidrepeat im Fratraxin Gen führt zu verminderter Expression von Fratraxin. Durchbruch in Hefe: Hefe YFH1 mutanten können nicht auf nicht fermentierbaren Kohlenstoffquellen wachsen, haben also defekte in der Atmungskette und sind sensitiv gegen H2O2. Frantraxin und YFH1 lokalisieren an Mitochondrien. Eisen Akkumuliert in YFH1 Mutanten und führt über Fenton zu ROS und Mitoschaden. Das konnte an Patienten bestätigt werden.

22
Friedreich`s Ataxia (FRDA)
Lokalisation von YFH in Hefe: Porin YFH-GFP Mito Tracker Frataxin Merge GFP

23
Eisenkonzentration steigt
Friedreich`s Ataxia (FRDA) Funktionsaufklärung in Hefe: Eisenkonzentration steigt AFT1-1up überexprimiert einen high affinity Eisentransporter.
 Funktionsaufklärung in Hefe: Eisenkonzentration steigt. AFT1-1up überexprimiert einen high. affinity Eisentransporter.")
24
Batten Disease -Blindheit, motorische Störungen, Tod im Kindesalter. Assoziiert mit Mutation in CLN3, einem unbekannten Protein. Akkumulierung von Proteinaggregaten in Lysosomen: Lipofuscin, ein autofluoreszentes Aggregat, dessen Hauptproteinkomponente aus der mitochondrialen ATPase stammt (Neuronale Lipofusceinosen). An Hefehomolog BTN1 wurde gezeigt: BTN1 ist wichtig für die Azidifizierung der Vakuole. Und lokalisiert auch dort. Kann durch humanes Homolog komplementiert werden, nicht jedoch durch krankmachende humane Mutanten. Dies ist eine Bühne für den funktionellen Test von BTN1 Mutanten.
. An Hefehomolog BTN1 wurde gezeigt: BTN1 ist wichtig für die Azidifizierung der Vakuole. Und lokalisiert auch dort. Kann durch humanes Homolog komplementiert werden, nicht jedoch durch krankmachende humane Mutanten. Dies ist eine Bühne für den funktionellen Test von BTN1 Mutanten.")
25
Frontemporale Demenz:
Atrophie des Stirn oder Schläfenlappens in Abwesenheit artheriosklerotischer oder Alzheimer-Phänotypen. Intellektueller Abbau, Veränderung der Persönlichkeit und des sozialen Verhaltens: -Einschlusskörper (Picksche Körper) -Frühe Symptome: Hang zu schmutzigen Witzen, sprachliche Enthemmung, Echolalie. Lebenserwartung: 5-10 Jahre Diagnose: PET
 -Frühe Symptome: Hang zu schmutzigen Witzen, sprachliche Enthemmung, Echolalie. Lebenserwartung: 5-10 Jahre. Diagnose: PET.")
26
-Reguliert Neurodegeneration in Drosphila
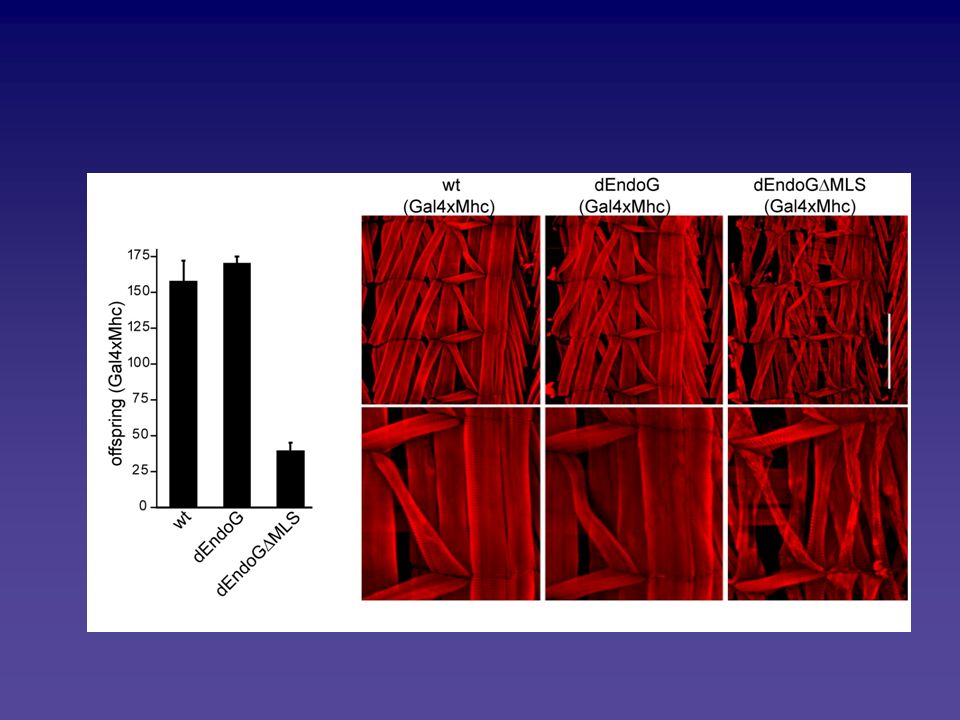
Frontemporale Demenz: VCP (CDC48) und Neurodegeneration VCP Colokalisiert mit Proteinaggregaten in Alzheimer’s, Parkinson’s, Huntington’s und Machado-Joseph diseases. Interagiert mit PolyQ in Vivo und in Vitro -Reguliert Neurodegeneration in Drosphila -Neuronale Zellkulturen, die PolyQ oder mutiertes VCP exprimieren zeigen Vesikelbildung und apoptotischen Zelltod. -Mitos aus Zellkulturen, die PolyQ exprimieren, sind an der perinuclearen Region assoziiert. CDC48 Mutante in Hefe löst Apoptose, ER Expansion und Vesikelbildung aus. Der Mechanismus konnte in Hefe erhellt werden.
 und Neurodegeneration. VCP Colokalisiert mit Proteinaggregaten in Alzheimer’s, Parkinson’s, Huntington’s und Machado-Joseph diseases. Interagiert mit PolyQ in Vivo und in Vitro. -Reguliert Neurodegeneration in Drosphila. -Neuronale Zellkulturen, die PolyQ oder mutiertes VCP exprimieren zeigen Vesikelbildung und apoptotischen Zelltod. -Mitos aus Zellkulturen, die PolyQ exprimieren, sind an der perinuclearen Region assoziiert. CDC48 Mutante in Hefe löst Apoptose, ER Expansion und Vesikelbildung aus. Der Mechanismus konnte in Hefe erhellt werden.")
27
VCP (CDC48) und Neurodegeneration
2D-Analysen zeigen, daß in der CDC48-Mutante lediglich die Mitochondrien dereguliert sind. Man stellt starke Bindung zwischen den Mitos und ER fest, ein Marker für Neurodegeneration

28
Figure 3 Braun, Zischka et al.

29
Figure 3 Braun, Zischka et al.

30
Free flow electrophorese

31
Figure 5 Braun, Zischka et al.

32
Figure 6 Braun, Zischka et al.

33
Parkinson

34
Absterben dopaminerger Neuronen
Beeinträchtigung der Motorik Hereditär und nicht-Hereditär

35
1817: Erste formale Beschreibung

36
Epidemiologie 1-2 % Betroffene bei den über 65jährigen
2.häufigste neurologische Erkrankung 1-2 % Betroffene bei den über 65jährigen Durchschnittsalter bei Krankheitsbeginn: 64,4 Jahre

37
Krankheitsverlauf

38
Symptome Rigor Tremor Weitere Symptome

39
Diagnose Klinische Kriterien
Physische Untersuchung und Erhebung der Patientengeschichte Fehldiagnoserate %

40
Ursache? Risikofaktoren Alter Familiäre Vorbelastung
Umwelteinflüsse (Schadstoffe, Umweltgifte, Mangan) Oxidativer Stress
 Oxidativer Stress.")
41
Molekularer Mechanismus
Lewy bodies Bestehend aus alpha-Synuclein, Ubiquitin, Neurofilamenten und anderen Proteinablagerungen

42
Alpha-Synuclein 140 AS langes präsynaptisches Protein
Parkinson-auslösende Mutationen: A53T, A30P und E46K sowie Gendupli- und triplikationen Wahrscheinlich am Proteintransport beteiligt Führt bei Parkinson-Kranken zu Störung des Vesikeltransports

43
Zelltod Dopaminsynthese im Cytoplasma
Dopamin neigt zur Erzeugung von ROS Oxidative Schäden > Zelltod Mitochondrienfunktion essentiell

44
Weitere mögliche Auslöser des Zelltods

45
Behandlung Erfolgt symptomatisch Dopaminagonisten
Hauptsächlich L-Dopa (Vorstufe von Dopamin) Tiefe Hirnstimulation
 Tiefe Hirnstimulation.")
46
Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models.

47
Plasmid overexpression screen identifies ER-Golgi trafficking genes as modifiers of αSyn toxicity
spotting assay shows that overexpression of ER-Golgi trafficking genes YPT1, YKT6, BRE5, UBP3, and ERV29 suppress αSyn-induced toxicity, whereas GYP8 and PMR1 overexpression enhances toxicity

48
Überexpression von YPT1 löst den Synuclein vermittelten Block es ER-Golgi transports

49
Überexpression von YPT1 rettet Würmer und Fliegen vor Parkinson

50
Conclusio The earliest defect following αSyn expression in yeast was a block in endoplasmic reticulum (ER) to Golgi vesicular trafficking. In a genomewide screen, the largest class of toxicity modifiers were proteins functioning at this same step, including the Rab guanosine triphosphatase Ypt1p, which associated with cytoplasmic αSyn inclusions. Elevated expression of Rab1, the mammalian YPT1 homolog, protected against αSyn-induced dopaminergic neuron loss in animal models of PD. Thus, synucleinopathies may result from disruptions in basic cellular functions that interface with the unique biology of particular neurons to make them especially vulnerable.

51
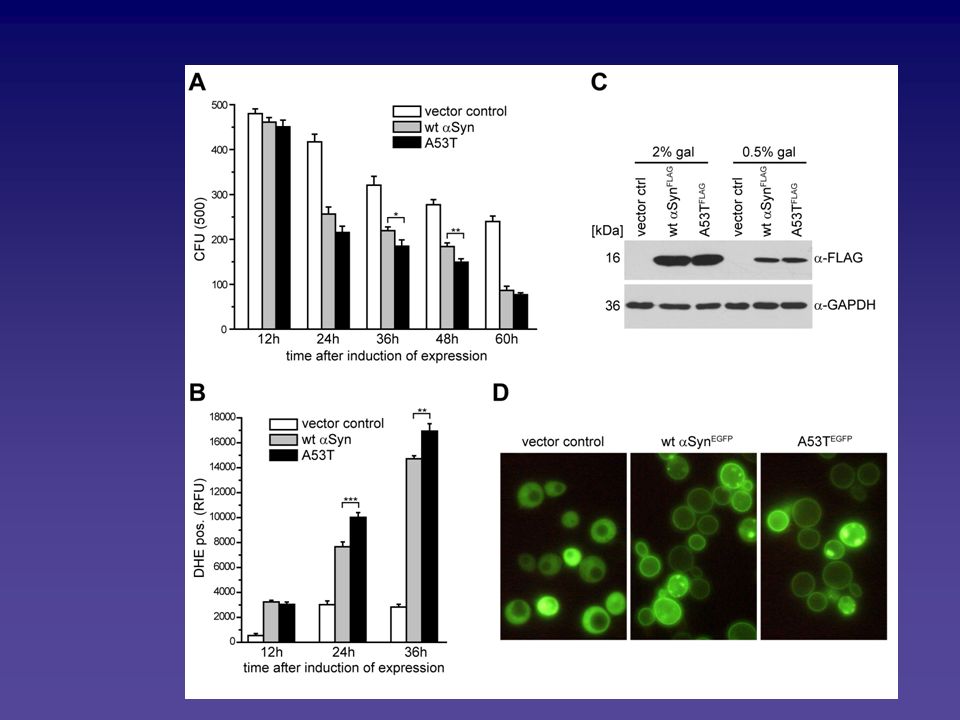
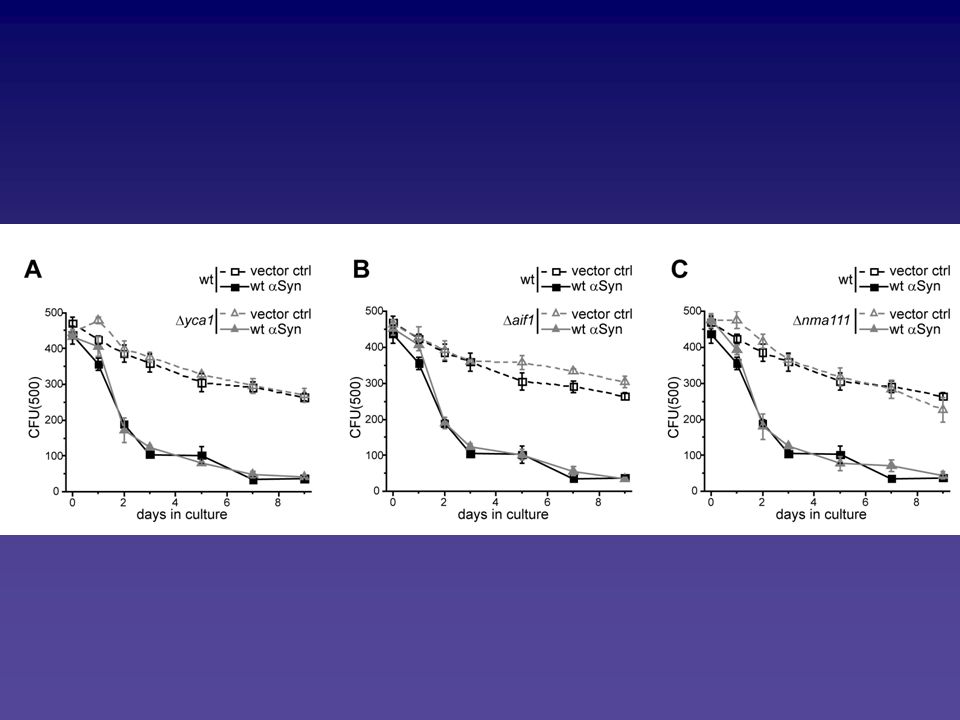
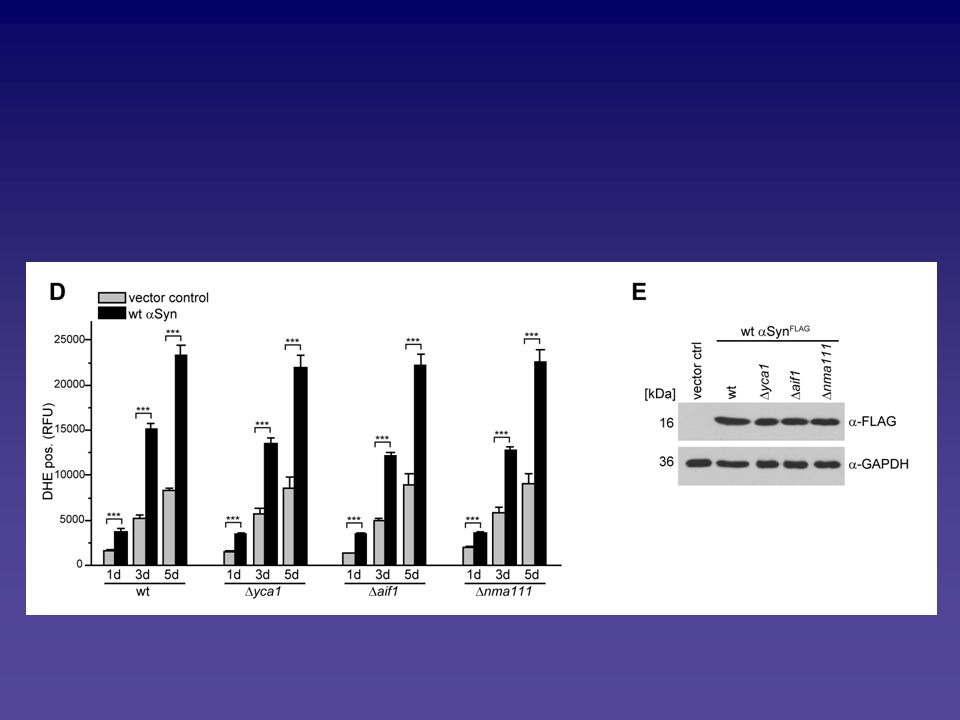
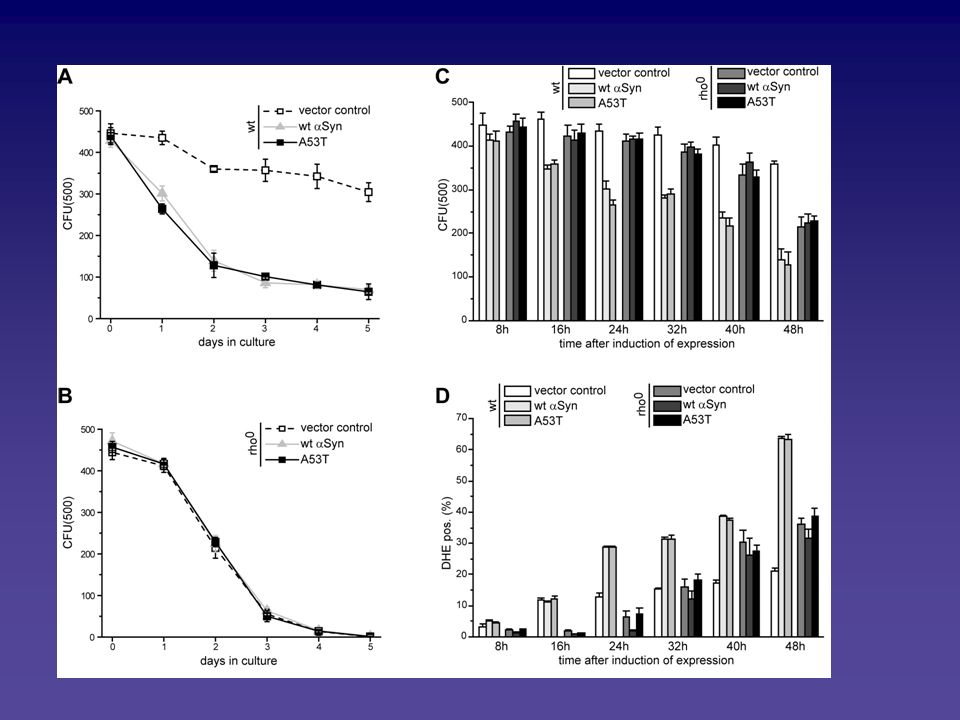
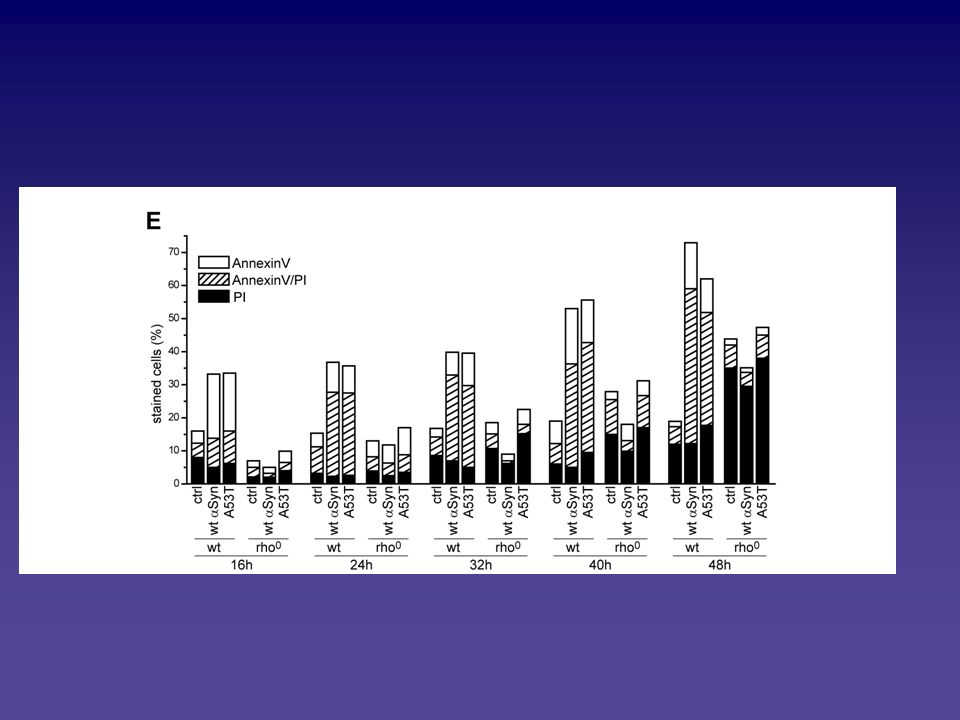
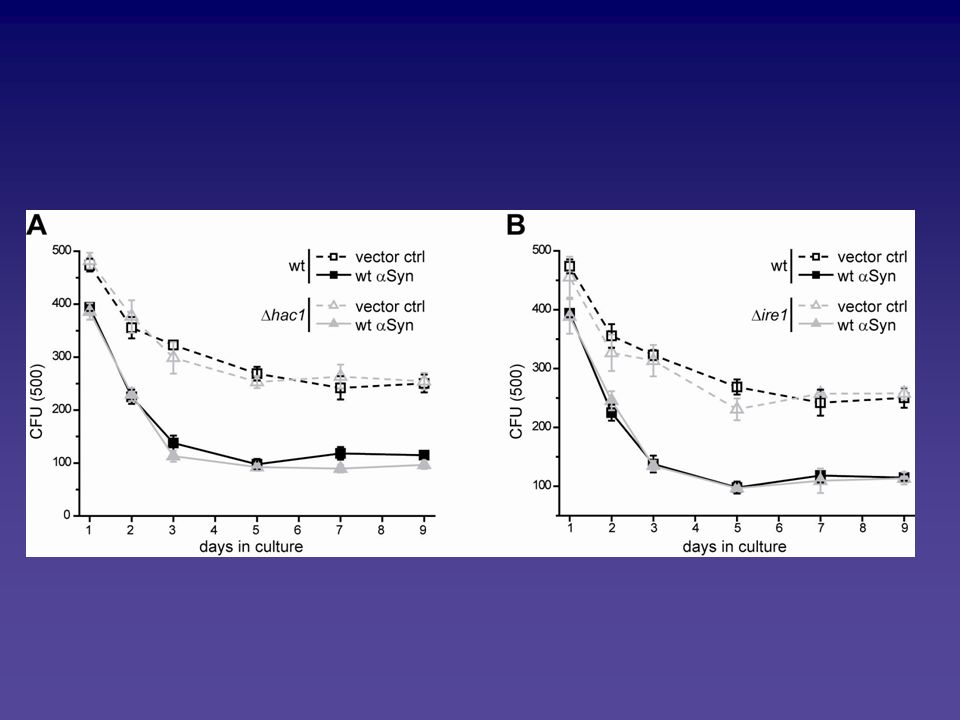
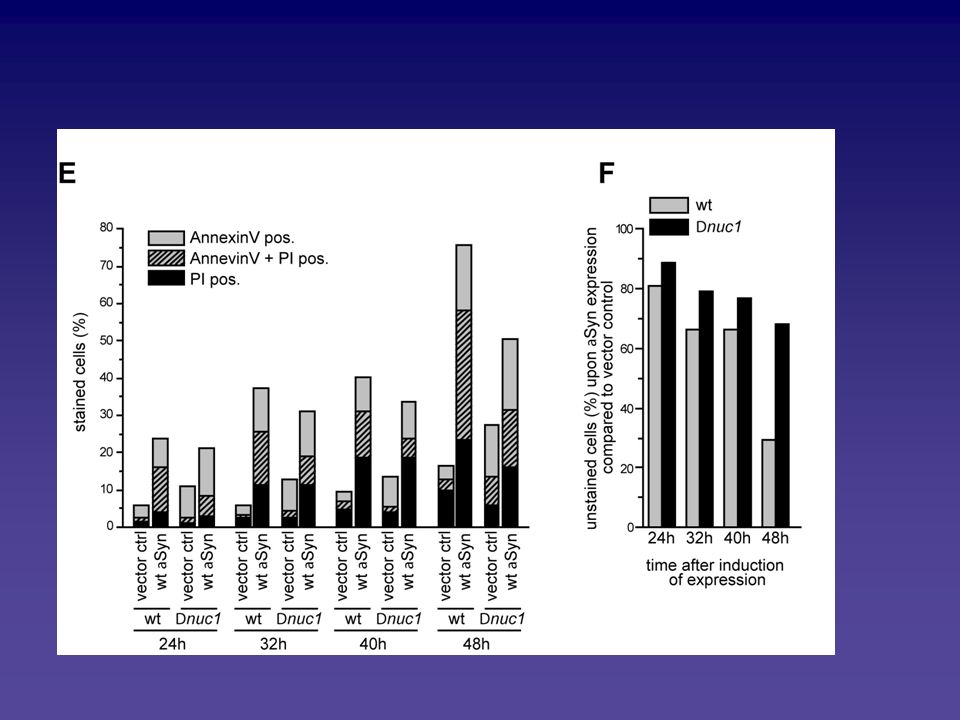
Analyse des Alpha-Synuclein vermittelten Zelltodphänotyps

64
Garvan Institute of Medical Research, Sydney
α-Synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity Received 1. October 2008; Accepted 14. November 2008; Published online 1. Feb. 2009 Aron D Gitler, Alessandra Chesi, Melissa L Geddie, Katherine E Strathearn, Shusei Hamamichi, Kathryn J Hill, Kim A Caldwell, Guy A Caldwell, Antony A Cooper, Jean-Christophe Rochet & Susan Lindquist Whitehead Institute for Biomedical Research and Howard Hughes Medical Institute, Cambridge Department of Cell and Developmental Biology, University of Pennsylvania School of Medicine, Philadelphia Department of Medicinal Chemistry and Molecular Pharmacology, Purdue University, West Lafayette Department of Biological Sciences, The University of Alabama, Tuscaloosa Garvan Institute of Medical Research, Sydney

65
Mangan-Exposition Mit PD in Verbindung stehender Umweltrisikofaktor
Zusammenspiel zwischen genetischem - und Umwelt-Risiko (PARK9 und Mangan)
")
66
Hefemodell Gene mit Funktion im Vesikel-Trafficking zwischen ER und Golgi verändern α-syn-Toxizität in Hefe Rab1: Säuger-ortholog von Ypt1 In neuronalem PD-Modell getestet Verhindert dopaminergen Neuronenverlust Screen mit 5000 weiteren Hefegenen Zusätzlich zu Genen für Vesikel-Trafficking auch einige weitere α-syn-Toxizität-beeinflussende Gene identifiziert Einige mit bekannten humanen Orthologen

67
Interaktionen Interaktionen zwischen den mit PD in Verbindung stehenden Loci Genetische Interaktionen zwischen PD-assoziierten Genen parkin und pink1 (Drosophila) α-syn und DJ-1 α-syn und ATP13A2 ATP13A2: lysosomale transmembrane Kation-transportierende P-Typ ATPase Verantwortlich für die frühe Form des Parkinsonismus mit pyramidaler Degeneration und Demenz (Kufor-Rakeb Syndrom)
 α-syn und DJ-1. α-syn und ATP13A2. ATP13A2: lysosomale transmembrane Kation-transportierende P-Typ ATPase. Verantwortlich für die frühe Form des Parkinsonismus mit pyramidaler Degeneration und Demenz (Kufor-Rakeb Syndrom)")
68
Hefehomolog von ATP13A2 unterdrückt α-syn-Toxizität
Suche nach Modifiern der α-syn-Toxizität YOR291W YPK9 (Yeast PARK9) Spotting assay: Mit α-syn-Toxizität Modifiern der Hefe YPT1 und YPK9 Kontrolle: Leervektor Glucose: α-syn-Expression unterdrückt Galactose: α-syn-Expression induziert
 Spotting assay: Mit α-syn-Toxizität Modifiern der Hefe. YPT1 und YPK9. Kontrolle: Leervektor. Glucose: α-syn-Expression unterdrückt. Galactose: α-syn-Expression induziert.")
69
Ypk9 und Ypt1 sind unabhängig
Beide starke Suppressoren der α-syn-Toxizität Ypt1: Hefehomolog zum humanen RAB1A GTPase reguliert Vesikel-Trafficking zwischen ER und Golgi Überexpression von Ypt1: verhindert α-syn-Toxizität HiTox-Stamm: exprimiert mehr α-syn und zeigt entsprechend höhere Toxizität Detektion von synergistischen Effekten zwischen verschiedenen Suppressoren Expression von Ypt1 oder Ypk9 alleine: nicht ausreichend um α-syn-Toxizität zu verhindern Coexpression ermöglicht Wachstum, aber keine komplette Unterdrückung der α-syn-Toxizität Genetische Interaktion zeigt Synergismus: Hinweis auf Funktion in verschiedenen Wegen

70
YPK9 Überexpresion schmilzt Synuclein Aggregate

71
Defekt im ER-Golgi-Trafficking
Carboxypeptidase Y (CPY) Subzellulare Lokalisation von CPY einfach zu bestimmen kompartiment-spezifische Glykosylierungen und proteolytische Spaltungen verändern die molekulare Masse des Proteins α-syn hindert CPY am verlassen des ER Überexpression von Ypt1 oder Ypk9 erlaubt Trafficking zum Golgi (weniger Protein im ER)
 Subzellulare Lokalisation von CPY einfach zu bestimmen. kompartiment-spezifische Glykosylierungen und proteolytische Spaltungen verändern die molekulare Masse des Proteins. α-syn hindert CPY am verlassen des ER. Überexpression von Ypt1 oder Ypk9 erlaubt Trafficking zum Golgi (weniger Protein im ER)")
72
Überexpression von Wildtyp Ypk9:
Unterdrückt α-syn-Toxizität Mutierte Proteine Beeinflussen α-syn-Toxizität nicht Beeinflussen Wachstum nicht Vakuoelenlokalisation und ATPase-Aktivität notwendig für Verhinderung der α-syn-Toxizität

73
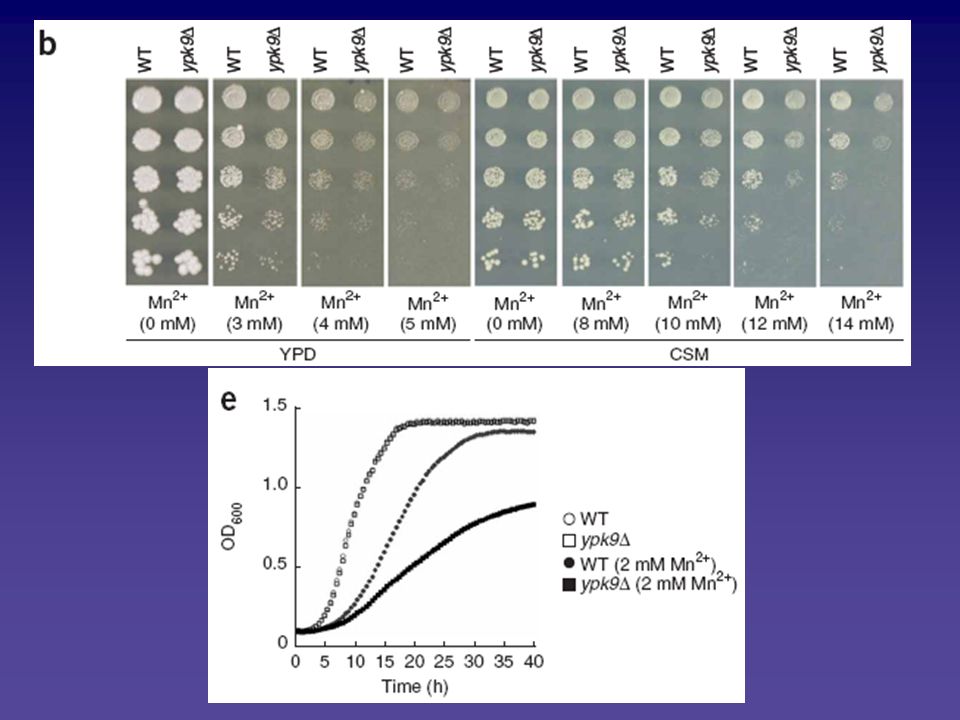
Mangan-Toxizität PARK9 und YPK9: transmembrane Kationen Metall-Transporter Wildtyp- und ypk9Δ-Zellen auf verschiedene Metalle getestet, um Substrat zu identifizieren ypk9Δ-Zellen sind hypersensitiv gegen Mn2+

75
Hefe-Ypk9 (und humanes ATP13A2/PARK9): Mangan-Transporter
Expression von Ypk9 von extrachromosomalem Plasmid mit starkem Promoter: Hebt Mn2+-Sensitivität wieder auf Macht ypk9Δ-Zellen und Wildtyp-Zellen resistenter gegen Mn2+ Hefe-Ypk9 (und humanes ATP13A2/PARK9): Mangan-Transporter
: Mangan-Transporter.")
76
Bestätigung in PD-Modellen
Ansätze für neue therapeutische Strategien 5 Suppressor-Gene getestet Ratten Neuron-Lentivirus-Modell C. elegans α-syn-Modell 4 davon wirksam PLK2 und PDE9A unterdrücken den α-syn-induzierten dopaminergen Neuronenverlust auch bei Nematoden

77
Conclusio Parkinson und Mangan
Manganese exposure is an environmental risk factor linked to PD and PD-like syndromes. Yeast PARK9 helps to protect cells from manganese toxicity, revealing a connection between PD genetics (alpha-syn and PARK9) and an environmental risk factor (PARK9 and manganese). Dopaminergic neuron loss caused by alpha-syn overexpression in animal and neuronal PD models is rescued by coexpression of PARK9. Further, knockdown of the Park9 ortholog in Caenorhabditis elegans enhances alpha-syn misfolding.
 and an environmental risk factor (PARK9 and manganese). Dopaminergic neuron loss caused by alpha-syn overexpression in animal and neuronal PD models is rescued by coexpression of PARK9. Further, knockdown of the Park9 ortholog in Caenorhabditis elegans enhances alpha-syn misfolding.")
78
Genetic Dissection of Spartin mediated Neurotoxicity in Yeast (Troyer syndrome, spastic paraplegia)
")
Ähnliche Präsentationen









 und der cytoplasmatischen Tyrosinkinase c-src in Urothelkarzinomzellen A. Melchior, J. Herrmann,>")













