Präsentation herunterladen
Die Präsentation wird geladen. Bitte warten

1
Reaktionsmechanismen in der (Anorganischen) Chemie
Theoretische Aspekte und Reaktionsmechanismen in der (Anorganischen) Chemie Theoretische Aspekte in der Anorganischen Chemie Peter Burger
 Chemie. Theoretische Aspekte. in der. Anorganischen Chemie. Peter Burger.")
2
Literatur Qualitative MO-Theorie:
- T. Albright et al. Orbital Interactions in Chemistry, Wiley 1985 - Y. Jean Molecular Orbitals of Transition Metal Complexes, Oxford University Press 2005 - Skript: T. Albright (meine Homepage) Reaktionsmechanismen: - R. Jordan, 3. Auflage, Reaction Mechanisms of Inorganic and Organo- metallic Systems, Wiley 2007 - E.V. Anslyn et al. Modern Physical Organic Chemistry University Science Books, 2006 Rechenverfahren & -methoden: - D. Young, Computational Chemistry: A Practical Guide for Applying Techniques to Real World Problems, Wiley 2001 (hier & heute) - E. Lewars Computational Chemistry, Kluwer F.H. Jensen, 2. Auflage, Introduction to Computational Chemistry, Wiley, 2006 - C. J. Cramer, 2. Auflage, Essentials of Computational Chemistry: Theories and Models Wiley 2004
 Reaktionsmechanismen: - R. Jordan, 3. Auflage, Reaction Mechanisms of Inorganic and Organo- metallic Systems, Wiley E.V. Anslyn et al. Modern Physical Organic Chemistry. University Science Books, Rechenverfahren & -methoden: - D. Young, Computational Chemistry: A Practical Guide for Applying Techniques to Real World Problems, Wiley 2001 (hier & heute) - E. Lewars Computational Chemistry, Kluwer F.H. Jensen, 2. Auflage, Introduction to Computational Chemistry, Wiley, C. J. Cramer, 2. Auflage, Essentials of Computational Chemistry: Theories and Models Wiley")
3
ausleihbar/verfügbar in der Chemiebibliothek
Literatur Qualitative MO-Theorie: - T. Albright et al. Orbital Interactions in Chemistry, Wiley 1985 - Y. Jean Molecular Orbitals of Transition Metal Complexes, Oxford University Press 2005 - Skript: T. Albright (meine Homepage) Reaktionsmechanismen: - R. Jordan, 3. Auflage, Reaction Mechanisms of Inorganic and Organo- metallic Systems, Wiley 2007 - E.V. Anslyn et al. Modern Physical Organic Chemistry University Science Books, 2006 Rechenverfahren & -methoden: - D. Young, Computational Chemistry: A Practical Guide for Applying Techniques to Real World Problems, Wiley 2001 (hier & heute) - E. Lewars Computational Chemistry, Kluwer F.H. Jensen, 2. Auflage, Introduction to Computational Chemistry, Wiley, 2006 - C. J. Cramer, 2. Auflage, Essentials of Computational Chemistry: Theories and Models Wiley 2004 ausleihbar/verfügbar in der Chemiebibliothek Altes/neues Skript & more: username: material: password: nitrogen
 Reaktionsmechanismen: - R. Jordan, 3. Auflage, Reaction Mechanisms of Inorganic and Organo- metallic Systems, Wiley E.V. Anslyn et al. Modern Physical Organic Chemistry. University Science Books, Rechenverfahren & -methoden: - D. Young, Computational Chemistry: A Practical Guide for Applying Techniques to Real World Problems, Wiley 2001 (hier & heute) - E. Lewars Computational Chemistry, Kluwer F.H. Jensen, 2. Auflage, Introduction to Computational Chemistry, Wiley, C. J. Cramer, 2. Auflage, Essentials of Computational Chemistry: Theories and Models Wiley ausleihbar/verfügbar in der Chemiebibliothek. Altes/neues Skript & more: username: material: password: nitrogen.")
4
ausleihbar/verfügbar in der Chemiebibliothek
Literatur Qualitative MO-Theorie: - T. Albright et al. Orbital Interactions in Chemistry, Wiley 1985 - Y. Jean Molecular Orbitals of Transition Metal Complexes, Oxford University Press 2005 - Skript: T. Albright (meine Homepage) Reaktionsmechanismen: - R. Jordan, 3. Auflage, Reaction Mechanisms of Inorganic and Organo- metallic Systems, Wiley 2007 - E.V. Anslyn et al. Modern Physical Organic Chemistry University Science Books, 2006 Rechenverfahren & -methoden: - D. Young, Computational Chemistry: A Practical Guide for Applying Techniques to Real World Problems, Wiley 2001 (hier & heute) - E. Lewars Computational Chemistry, Kluwer F.H. Jensen, 2. Auflage, Introduction to Computational Chemistry, Wiley, 2006 - C. J. Cramer, 2. Auflage, Essentials of Computational Chemistry: Theories and Models Wiley 2004 ausleihbar/verfügbar in der Chemiebibliothek Altes/neues Skript & more: username: material: password: nitrogen
 Reaktionsmechanismen: - R. Jordan, 3. Auflage, Reaction Mechanisms of Inorganic and Organo- metallic Systems, Wiley E.V. Anslyn et al. Modern Physical Organic Chemistry. University Science Books, Rechenverfahren & -methoden: - D. Young, Computational Chemistry: A Practical Guide for Applying Techniques to Real World Problems, Wiley 2001 (hier & heute) - E. Lewars Computational Chemistry, Kluwer F.H. Jensen, 2. Auflage, Introduction to Computational Chemistry, Wiley, C. J. Cramer, 2. Auflage, Essentials of Computational Chemistry: Theories and Models Wiley ausleihbar/verfügbar in der Chemiebibliothek. Altes/neues Skript & more: username: material: password: nitrogen.")
6
Anorganische Mechanismen nur bis 10 zählen können

7
Elementarschritte

8
Elementarschritte

9
Elementarschritte

10
Beispiel - Katalyse: Kreuzkupplungsreaktion

11
Kreuzkupplungsreaktionen
[LnM] R-X + Nu R-Nu + X Katalyse ! bama.ua.edu/~kshaughn/ch609/notes/6-cross-couple.pdf Mn: Metallatome/ionen? - Haare spalten?

12
& mehr Details schon besser: LnMn

13
Katalysatoraktivierung & Nebenprodukte
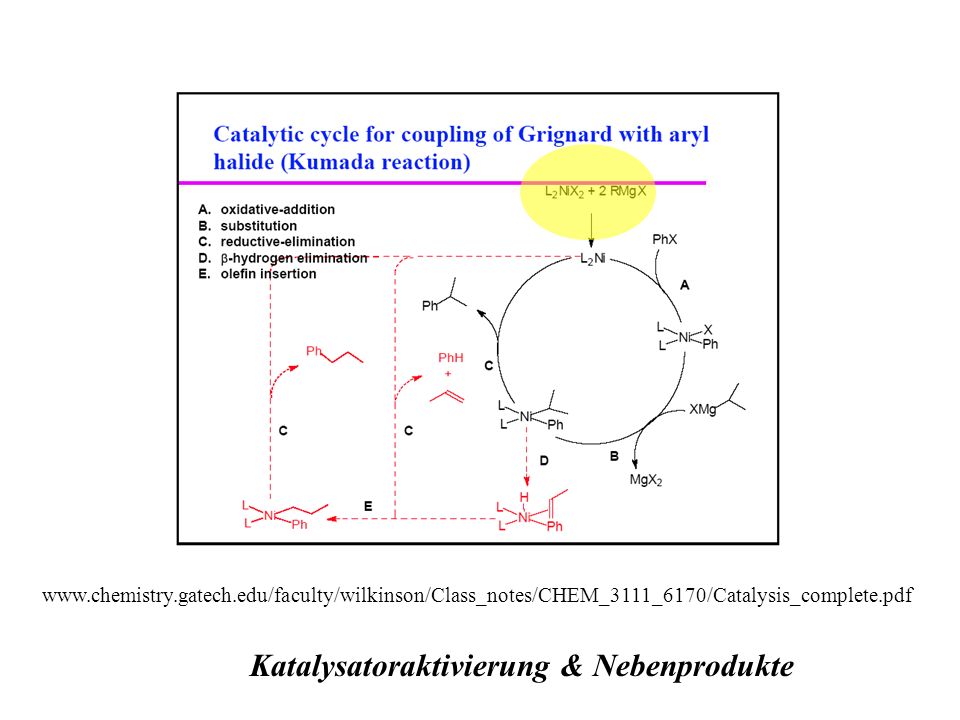
16
"Verbesserung" höhere Ausbeute in Gegenwart von
eletronenarmen Olefinen

17
Mechanismen lassen sich nicht beweisen!
Ni-vermittelte Aryl-Kupplung Papier ist willig!!!!!!!! Mechanismen lassen sich nicht beweisen!

18
keep it simple ! denn es kann alles auch viel komplizierter gehen
Occam's Razor: (William of Ockham) (Kybernetik) one should not increase, beyond what is necessary, the number of entities required to explain anything keep it simple !
 (Kybernetik) one should not increase, beyond what is necessary, the number of entities required to explain anything. keep it simple !")
19
Mechanismen kann man nur widerlegen
aber!: Mechanismen kann man nur widerlegen

20
Noch ein Vorschlag zum Mechanismus

21
C-C-Verknüpfung

22
Reduktive Eliminierung
Geschwindigkeitskonstante schnell/langsam? warum Unterschiede?

23
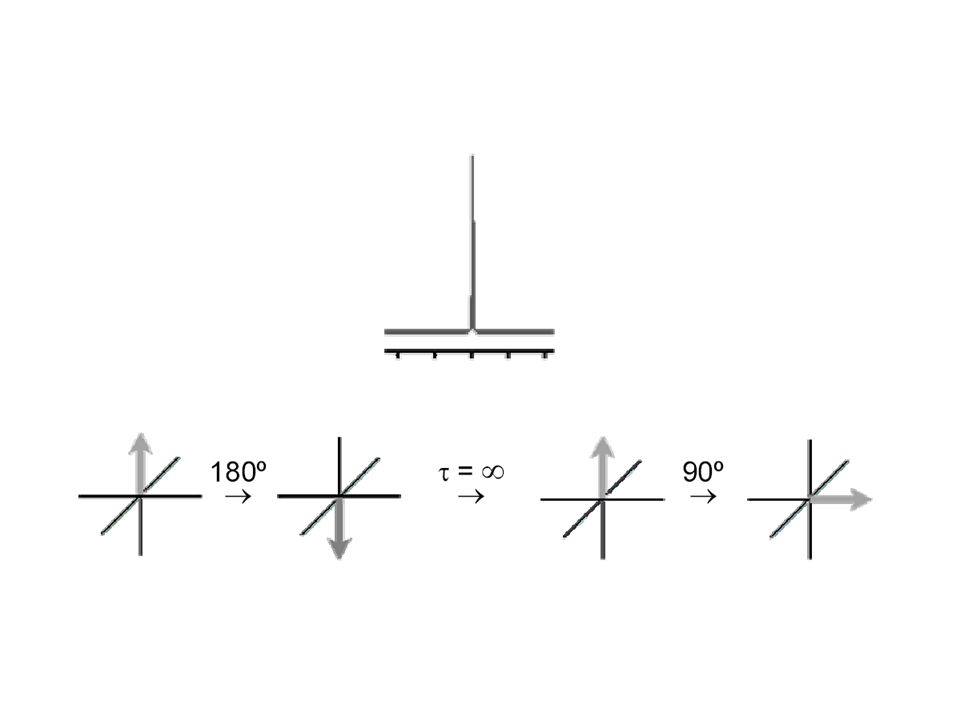
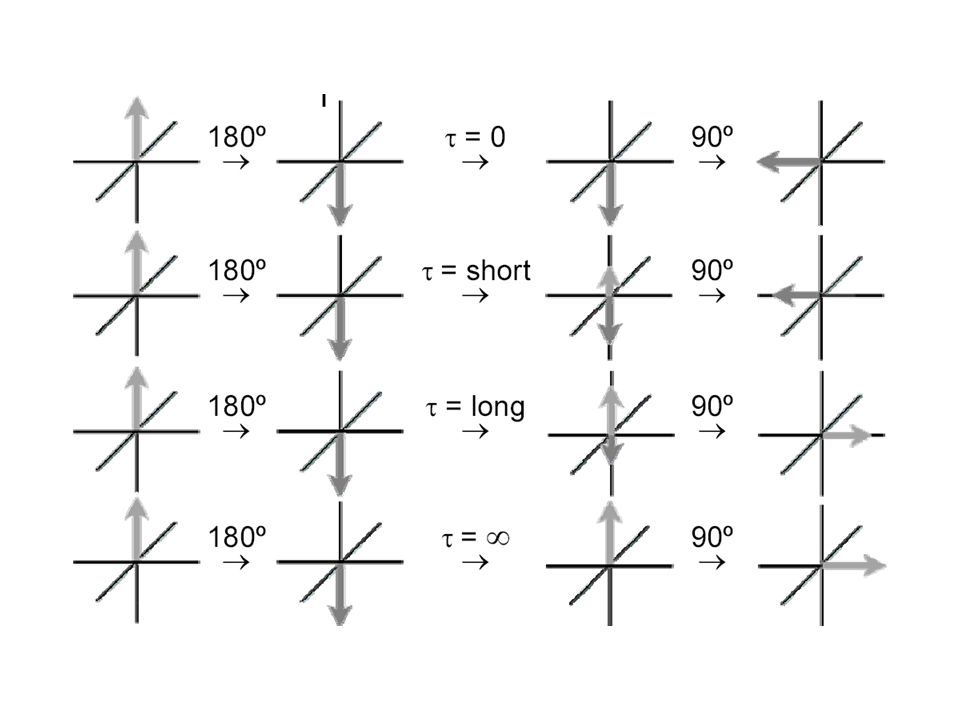
[ = [ ]o .e-kt [ = 1/2 [ ]o 1/2 = e-kt1/2 t1/2= ln 2/k t1/2 [sec]
´000 5 t1/2 5 t1/2: = 96.8 % Umsatz ~ 1 h /2 d Woche Katalyse ? stabil, inert? Reaktion 1. Ordnung [ (t)] = [ ]o .e-kt Halbwertszeit, t1/2: [ (t)] = 1/2 [ ]o 1/2 = e-kt1/2 t1/2= ln 2/k
![[ = [ ]o .e-kt [ = 1/2 [ ]o 1/2 = e-kt1/2 t1/2= ln 2/k t1/2 [sec]](http://slideplayer.org/slide/208366/1/images/23/%5B+%3D+%5B+%5Do+.e-kt+%5B+%3D+1%2F2+%5B+%5Do+1%2F2+%3D+e-kt1%2F2+t1%2F2%3D+ln+2%2Fk+t1%2F2+%5Bsec%5D.jpg "´ t1/2. 5 t1/2: = 96.8 % Umsatz. ~ 1 h 1/2 d 1 Woche. Katalyse stabil, inert Reaktion 1. Ordnung. [ (t)] = [ ]o .e-kt. Halbwertszeit, t1/2: [ (t)] = 1/2 [ ]o. 1/2 = e-kt1/2. t1/2= ln 2/k.")
24
stabil vs inert DG DGR < 0 instabil + r

25
stabil DGR > 0 DG + stabil Thermodynamik r

26
DGR vs K - van´t Hoff DG = -RT.lnK A B
DGR= 0 kcal/mol [A]/[B]= = " = 10 = " = 100

27
inert in/stabil vs inert Kinetik DG DG# groß !!!!langsam!!!! DG# r
+ r

28
Einschub Eyring-Gleichung (aktivierter Komplex)
kB = R/NA T: Temperatur h; Planck´sches Wirkungsquantum DG# freie Enthalpie der Aktivierung

30
DG DDG# r

31
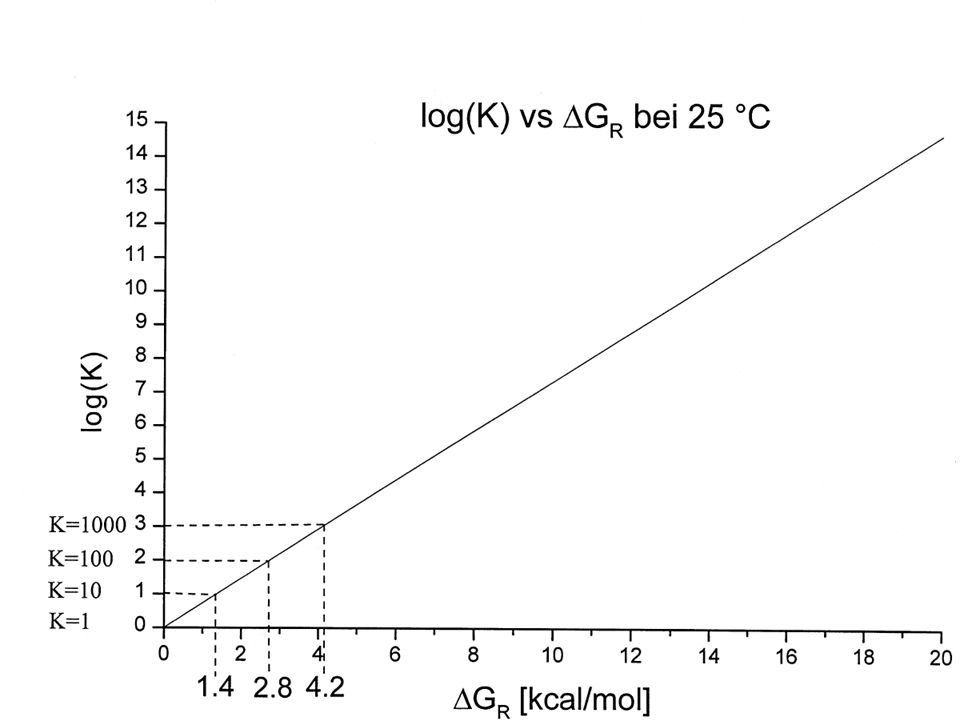
DDG# = 1.4 kcal/mol = 10x schneller

33
stabil Abschätzung Thermodynamik DG r
+ stabil r Gebrochene & neu gebildete Bindungen

34
- {(BDE(Me-Me) - 2 BDE(Pd-Me}
1) Gebrochene Bindungen: 2 BDE(Pd-Me) 2) Neugebildete Bindungen: 1 BDE(Pd-Me) DH ~ - {S BDE(neue Bdg.) - S BDE(gebr. Bdg)} ~ DH ~ ~ - {(BDE(Me-Me) - 2 BDE(Pd-Me} BDEs ?
 Gebrochene Bindungen: 2 BDE(Pd-Me) 2) Neugebildete Bindungen: 1 BDE(Pd-Me) DH ~ - {S BDE(neue Bdg.) - S BDE(gebr. Bdg)} ~ DH ~ ~ - {(BDE(Me-Me) - 2 BDE(Pd-Me} BDEs")
35
LnM-R,H Bindungsdissoziationsenthalpien - Trends
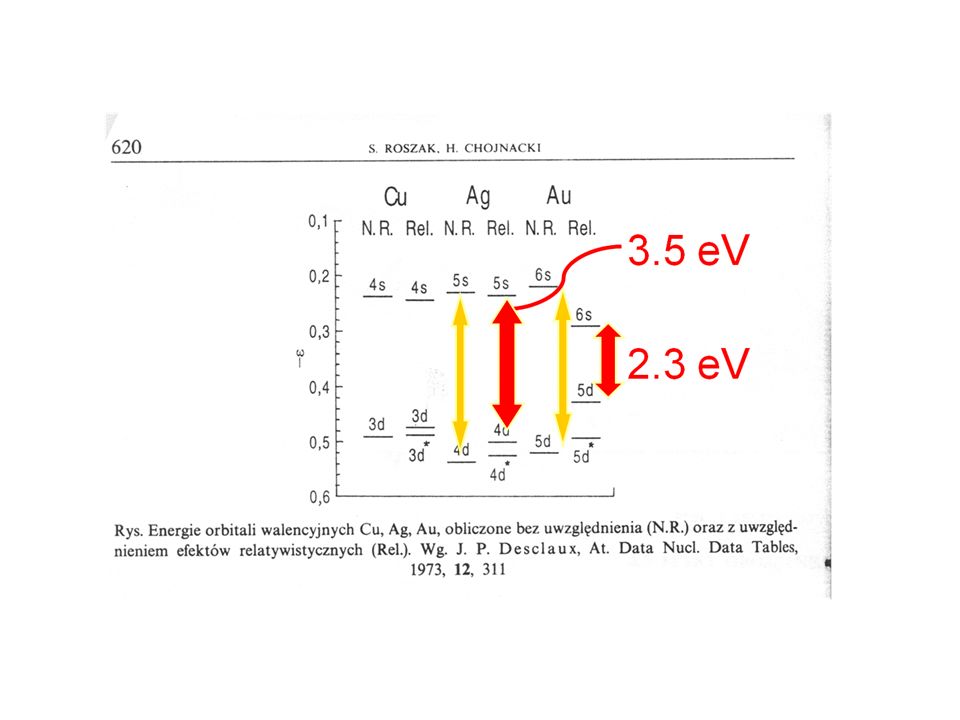
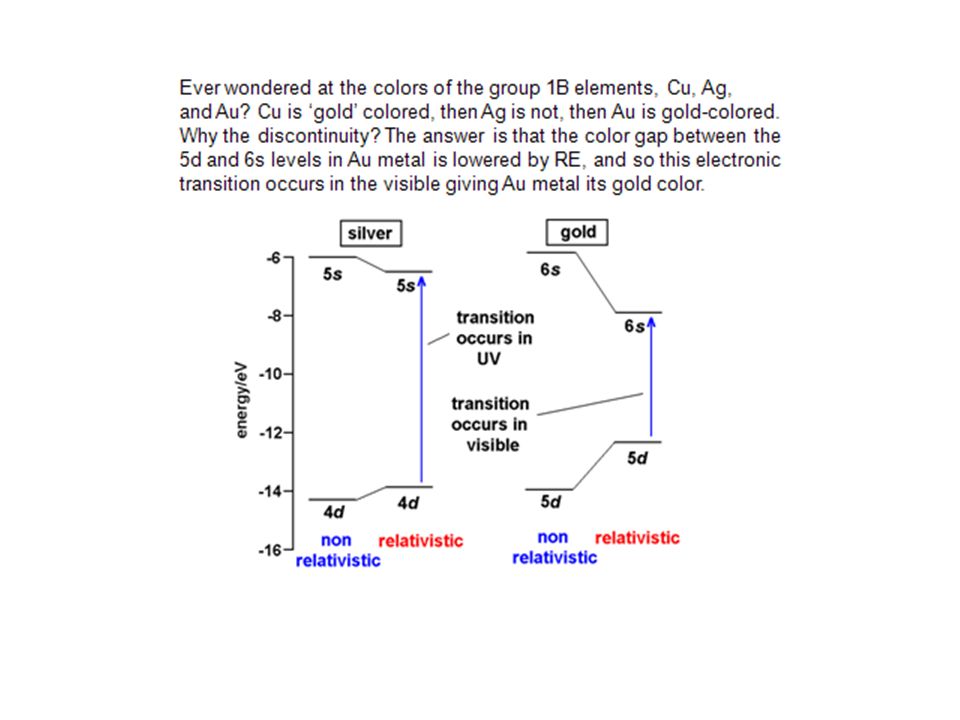
elektropositive Metalle: frühe Übergangsmetalle, Aktinoide Referenz für Th, U: D(Th,U)-O 123 bzw. 115 kcal/mol. Gruppe(IV) basierend auf D(M-Cl) 102, 117, 119. sehr starke M-C und M-H Bindungen, D(M-H) und D(M-alkyl) sehr ähnlich Reihenfolge Bindungsstabilität: 5d > 4d > 3d
-O 123 bzw. 115 kcal/mol. Gruppe(IV) basierend auf D(M-Cl) 102, 117, 119. sehr starke M-C und M-H Bindungen, D(M-H) und D(M-alkyl) sehr ähnlich. Reihenfolge Bindungsstabilität: 5d > 4d > 3d.")
36
LnM-R,H Bindungsdissoziationsenthalpien - Trends
mittlere - späte Übergangsmetalle Referenz für Mn, Re; D((CO)5M-M(CO)5) M-H Bindungen stärker als M-Alkyl Bindung: Differenz kcal/mol M-C-Bdg. für 3d-Metalle ziemlich schwach, für späte 5d-ÜM vergleichbar stark wie für frühe ÜM Reihenfolge Bindungsstabilität: 5d > 4d > 3d diffusere Orbitale für höhere Homologe => besserer Überlapp; 5d vs 4d: Relativistik (ca kcal/mol stab.)
5M-M(CO)5) M-H Bindungen stärker als M-Alkyl Bindung: Differenz kcal/mol. M-C-Bdg. für 3d-Metalle ziemlich schwach, für späte 5d-ÜM vergleichbar stark wie für frühe ÜM. Reihenfolge Bindungsstabilität: 5d > 4d > 3d. diffusere Orbitale für höhere Homologe => besserer Überlapp; 5d vs 4d: Relativistik (ca kcal/mol stab.)")
37
rel. BDE(LnRu-X) [kcal/mol]
Korrelation BDE(H-X) vs BDE(LnM-X) BDE(H-X) [kcal/mol] lineare Korrelation! aber Steigung < 1 H-X Bdg. stärker rel. BDE(LnRu-X) [kcal/mol]
![rel. BDE(LnRu-X) [kcal/mol]](http://slideplayer.org/slide/208366/1/images/37/rel.+BDE%28LnRu-X%29+%5Bkcal%2Fmol%5D.jpg "Korrelation BDE(H-X) vs BDE(LnM-X) BDE(H-X) [kcal/mol] lineare Korrelation! aber Steigung < 1. H-X Bdg. stärker. rel. BDE(LnRu-X) [kcal/mol]")
39
R3C-X Bindungsdissoziationsenthalpien
BDE + RnC-X RnC + X

40
? Bindungsstärke: A-B: Bindungsstärke
unpolar kovalent polar kovalent ionisch H-F d+ d- H2 Na+ Cl- Gasphase BDE: [kcal/mol] ? 104 135 95 A-B: Bindungsstärke (L. Pauling) großer EN-Unterschied stärkt Bdg. BDE(A-B)= ½{BDE(A-A)+BDE(B-B)} + C. (EN(A)-EN(B))2
 großer EN-Unterschied stärkt Bdg. BDE(A-B)= ½{BDE(A-A)+BDE(B-B)} + C. (EN(A)-EN(B))2.")
41
BDE Korrelationen H-X vs H3C-X
BDE(H-X) BDE(C-X) X
 BDE(C-X) X.")
42
D = BDE(H-X)- BDE(C-X) =
BDE Korrelationen H-X vs H3C-X BDE(A-B)= ½{BDE(A-A)+BDE(A-A)} + C. (EN(A)-EN(B))2 D = BDE(H-X)- BDE(C-X) = 2C. (EN(C)-EN(H)).EN(X) + ½{BDE(H-H)-BDE(CH3-CH3)} +C{EN(H)2-EN(C)2 )} D EN(X) D ~ EN(X) X
= ½{BDE(A-A)+BDE(A-A)} + C. (EN(A)-EN(B))2. D = BDE(H-X)- BDE(C-X) = 2C. (EN(C)-EN(H)).EN(X) + ½{BDE(H-H)-BDE(CH3-CH3)} +C{EN(H)2-EN(C)2 )} D. EN(X) D ~ EN(X) X.")
43
Reduktive Eliminierung
BDE(C-C)100 kcal/mol 2 BDE(M-C)100 kcal/mol DHR -{S BDE(neue Bdg.) - S BDE(gebr. Bdg.)} DHR = -(BDE(C-C) - 2 BDE(M-C)}
100 kcal/mol. 2 BDE(M-C)100 kcal/mol. DHR -{S BDE(neue Bdg.) - S BDE(gebr. Bdg.)} DHR = -(BDE(C-C) - 2 BDE(M-C)}")
44
LnM-Me Bindungsdissoziationsenthalpien - Trends
elektropositive Metalle: frühe Übergangsmetalle, Aktinoide Referenz für Th, U: D(Th,U)-O 123 bzw. 115 kcal/mol. Gruppe(IV) basierend auf D(M-Cl) 102, 117, 119.
-O 123 bzw. 115 kcal/mol. Gruppe(IV) basierend auf D(M-Cl) 102, 117, 119.")
45
LnM-Me Bindungsdissoziationsenthalpien - Trends
mittlere - späte Übergangsmetalle Referenz für Mn, Re; D((CO)5M-M(CO)5)
5M-M(CO)5)")
46
Reduktive Eliminierung
2 BDE(M-C)100 kcal/mol BDE(C-C)100 kcal/mol DHR -{S BDE(neue Bdg.) - S BDE(gebr. Bdg.)} DHR = -(BDE(C-C) - 2 BDE(M-C)} = -( · 70) frühes ÜM = -( · 50) spätes ÜM DHR = +40 kcal/mol frühes ÜM = " spätes ÜM Stabilität DG!!! nicht DH
100 kcal/mol. BDE(C-C)100 kcal/mol. DHR -{S BDE(neue Bdg.) - S BDE(gebr. Bdg.)} DHR = -(BDE(C-C) - 2 BDE(M-C)} = -( · 70) frühes ÜM. = -( · 50) spätes ÜM. DHR = +40 kcal/mol frühes ÜM. = 0 spätes ÜM. Stabilität DG!!! nicht DH.")
47
DS ? DG = DH - TDS Gas: DS 30 eu (entropy units) cal mol K-1 Merken!
bei RT: TDS = 300·30 = 9000 cal/mol 10 kcal/mol DHR = -40 kcal/mol frühes ÜM = " spätes ÜM DG = DH - TDS = = +30 kcal/mol frühes ÜM = = -10 kcal/mol spätes ÜM Pd-Dialkyl instabil!!! aber isolierbar => inert!!

48
inert instabil & inert Kinetik DG DG# groß DG# K298? r
+ DGR = -10 kcal/mol instabil K298? r

49
K = 107 DGR vs K - van´t Hoff DG = -RT.lnK A B
DGR= 0 kcal/mol [A]/[B]= = " = 10 = " = 100 A B K = 107

50
Kalorimeter DHR Messung
relative/absolute Bindungsstärken - thermochemische Titration Kalorimeter DHR Messung BDE´s ? DHR -{S BDE(neue Bdg.) - S BDE(gebr. Bdg.)}
 - S BDE(gebr. Bdg.)}")
51
DHR -{S BDE(neue Bdg.) - S BDE(gebr. Bdg.)}
DHR -{2 BDE(Th-OtBu) - 2 BDE(Th-R) -2 BDE(tBu-OH)} BDE(Th-R) -BDE(Th-OtBu) - ½ DHR BDE(Th-R´s) gemittelt! Wasser: BDE1(H2O HO· + H·) = 120 kcal/mol BDE2(HO· O + H·) = 100 kcal/mol BDE(H2O) = 110 kcal/mol
 - 2 BDE(Th-R) -2 BDE(tBu-OH)} BDE(Th-R) -BDE(Th-OtBu) - ½ DHR. BDE(Th-R´s) gemittelt! Wasser: BDE1(H2O HO· + H·) = 120 kcal/mol. BDE2(HO· O + H·) = 100 kcal/mol. BDE(H2O) = 110 kcal/mol.")
52
eigentlich etwas mehr PC..
DHR(g) = Solvatationsenthalpie
 = Solvatationsenthalpie.")
53
Solvatationsenthalpien exp.

54
BDE(Th-OtBu) = 124 kcal/mol
BDE(Th-R,X) [kcal/mol] BDE(Th-R,X)solv [kcal/mol] R = Me 78(1) R = Et 71(2) R = Ph 92(2) R = H 90(1) R = Et, X = Cl 68(2) BDE(Th-OtBu) = 124 kcal/mol
 [kcal/mol] BDE(Th-R,X)solv. [kcal/mol] R = Me. 78(1) R = Et. 71(2) R = Ph. 92(2) R = H. 90(1) R = Et, X = Cl. 68(2) BDE(Th-OtBu) = 124 kcal/mol.")
55
Mittlere Bindungsdissoziationsenthalpien homoleptischer Verbindungen
BDE(M-Me)
")
56
Natur: Metallorganik - Coenzym B12
• + Homolyse

57
B12-Modellsysteme: BDE-Bestimmung aus Gleichgewichtsmessungen
R L + R• • K L = R = NH2, Me, H, CN aber exp.: K

58
DHR = DH1+ DH2 BDE(Co-R) • • Kexp
Themochemischer Zyklus / Umrechnung LnCo-CH(CH3)Ph LnCo• + CH2=CH-Ph + ½ H2 DH1 Kexp CH2=CH-Ph + ½ H2 CH3-CH-Ph DH02 • = -2.2 kcal/mol (Lit.) • LnCo-CH(CH3)Ph LnCo• + CH3-CH-Ph DHR = DH1+ DH2 BDE(Co-R)
Ph LnCo• + CH2=CH-Ph + ½ H2 DH1. Kexp. CH2=CH-Ph + ½ H2 CH3-CH-Ph DH02. • = -2.2 kcal/mol (Lit.) • LnCo-CH(CH3)Ph LnCo• + CH3-CH-Ph. DHR = DH1+ DH2 BDE(Co-R)")
59
Kexp Kexp Gleichgewichtsreaktion (UV/VIS-Messung)
LnCo-CH(CH3)Ph LnCo• + CH2=CH-Ph + ½ H2 DH1 Kexp Kexp
Ph LnCo• + CH2=CH-Ph + ½ H2 DH1. Kexp. Kexp.")
60
Thermodynamik: Temperaturabhängigkeit von K
van´t Hoff Auftragung ln K(T) 1/T 1/T lnK • Achsenabschnitt: Steigung: "gutes Experiment": DT mindestens 40 K
 1/T. 1/T. lnK. • Achsenabschnitt: Steigung: gutes Experiment : DT mindestens 40 K.")
61
L = R L L/X BDE(Co-R) [kcal/mol] 21.2 20.1 19.5 17.9 20.8
X = NH2, Me, H, CN L/X X = NH2 X = Me X = H X = CN L = BDE(Co-R) [kcal/mol] 21.2 20.1 19.5 17.9 20.8
![L = R L L/X BDE(Co-R) [kcal/mol]](http://slideplayer.org/slide/208366/1/images/61/L+%3D+R+L+L%2FX+BDE%28Co-R%29+%5Bkcal%2Fmol%5D.jpg "X = NH2, Me, H, CN. L/X. X = NH2. X = Me. X = H. X = CN. L = BDE(Co-R) [kcal/mol]")
62
k12, langsam (rds) Reaktion 1. Ordnung
B12-Modellsysteme: BDE-Bestimmung aus Kinetikdaten L(DH)2Co-R k12, langsam (rds) schnell schnell Reaktion 1. Ordnung
2Co-R. k12, langsam (rds) schnell. schnell. Reaktion 1. Ordnung.")
63
Reaktionsschema E k,DH#, DS# K, DGR DH# > BDE(Co-R) BDE(Co-R)
[LnCo]-H CH2=CHPh [LnCo•]...H3CHPh# {[LnCo•] •CH(CH3)Ph}# [LnCo•] + ½ H2 + CH2=CHPh K, DGR [LnCo]-H....H-[CoLn]# [LnCo•] + •CH(CH3)Ph BDE(Co-R) k,DH#, DS# [LnCo]-CH(CH3)Ph DH# > BDE(Co-R)
Ph}# [LnCo•] + ½ H2 + CH2=CHPh. K, DGR. [LnCo]-H....H-[CoLn]# [LnCo•] + •CH(CH3)Ph. BDE(Co-R) k,DH#, DS# [LnCo]-CH(CH3)Ph. DH# > BDE(Co-R)")
64
Kinetik: Temperaturabhängigkeit von k
ln(k/T) • Eyring-Gleichung Eyring Auftragung ln k/T 1/T Achsenabschnitt: Steigung: "gutes Experiment": DT mindestens 40 K
 • Eyring-Gleichung. Eyring Auftragung ln k/T 1/T. Achsenabschnitt: Steigung: gutes Experiment : DT mindestens 40 K.")
65
BDE(Co-R)
")
66
BDE´s Metallhydride / Elektrochemie / Chemie

67
Thermochemischer Zyklus
Acidität: Redox: BDE: BDEDG = 1.37 pKa Eoox(M-) [kcal/mol]
 [kcal/mol]")
68
BDEDG = 1.37 pKa + 23.06 Eoox(M-) + 53.6 [kcal/mol]
Vergleich mit kalorimetrischen Messungen (Entropieanteil) BDEDH = BDE = 1.37 pKa Eoox(M-) [kcal/mol]
![BDEDG = 1.37 pKa Eoox(M-) [kcal/mol]](http://slideplayer.org/slide/208366/1/images/68/BDEDG+%3D+1.37+pKa+Eoox%28M-%29+%5Bkcal%2Fmol%5D.jpg "Vergleich mit kalorimetrischen Messungen (Entropieanteil) BDEDH = BDE = 1.37 pKa Eoox(M-) [kcal/mol]")
69
Elektrochemie LnM- LnM· + e- E0ox 2 LnM· LnM_MLn (reversibel !)
Referenz: Cp2Fe+/Cp2Fe in CH3CN, kdim in [Msec-1], E´s in [V] kdim 2 LnM· LnM_MLn (irreversibel) Ecorr
 Ecorr.")
70
M-H Bindungsdissoziationsenthalpien
BDE(M-H) [kcal/mol]
 [kcal/mol]")
71
M-H Bindungsdissoziationsenthalpien
BDE(M-H) [kcal/mol] Lit.
 [kcal/mol] Lit.")
72
M-H Bindungsdissoziationsenthalpien
BDE(M-H) [kcal/mol] Eox Lit.
 [kcal/mol] Eox. Lit.")
73
M-H Bindungsdissoziationsenthalpien
BDE(M-H) [kcal/mol] Eox pKa Lit.
 [kcal/mol] Eox. pKa. Lit.")
74
Thermochemische Zyklen - weitere Anwendung
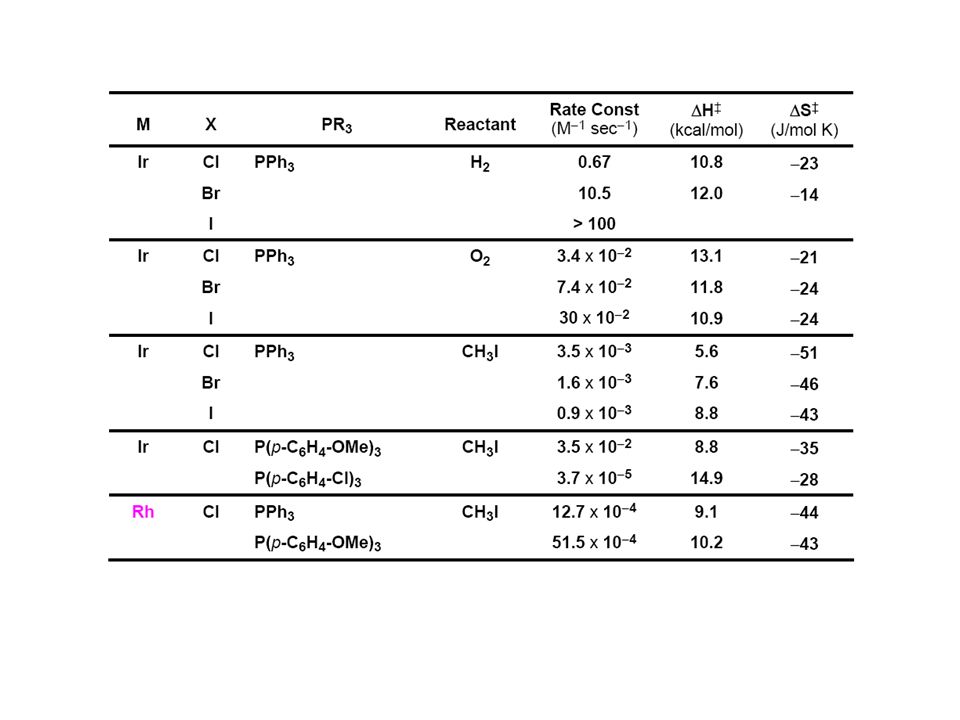
pKa(M-H)+·
+·")
75
pKa(M-H)+· DpKa -20 !!!
+· DpKa -20 !!!")
78
N2-Aktivierung bei RT! BDE(N-N) = 226 kcal/mol
 = 226 kcal/mol")
79
DHR: Messung von DHR im Kalorimeter

80
From somewhere in webspace....

81
65 Minuten und noch 140 Folien...
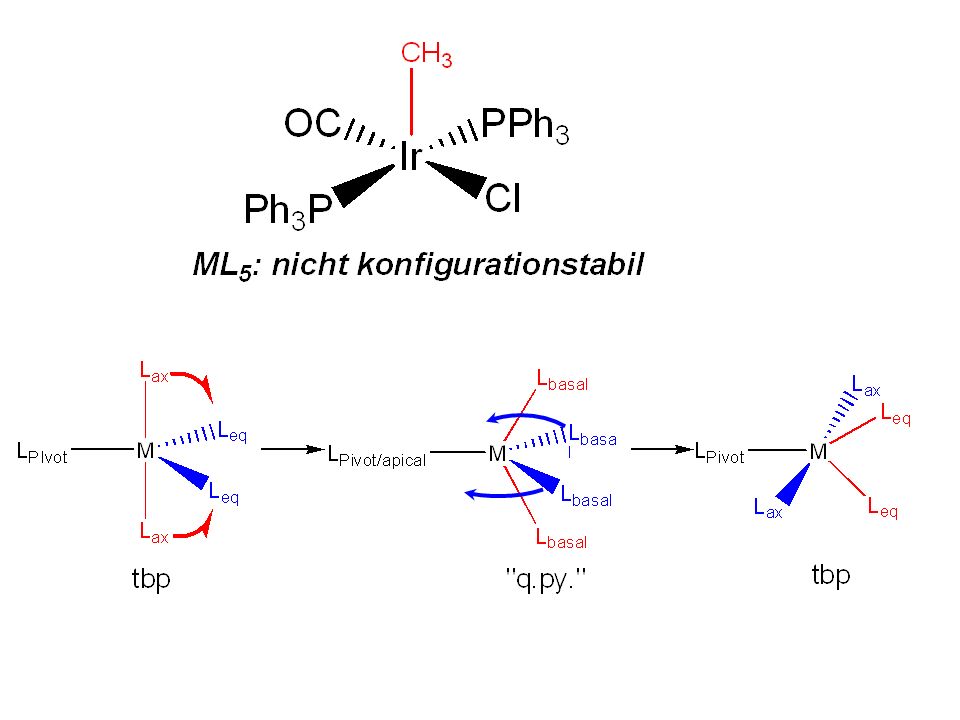
Metallorganik 11 65 Minuten und noch 140 Folien...

83
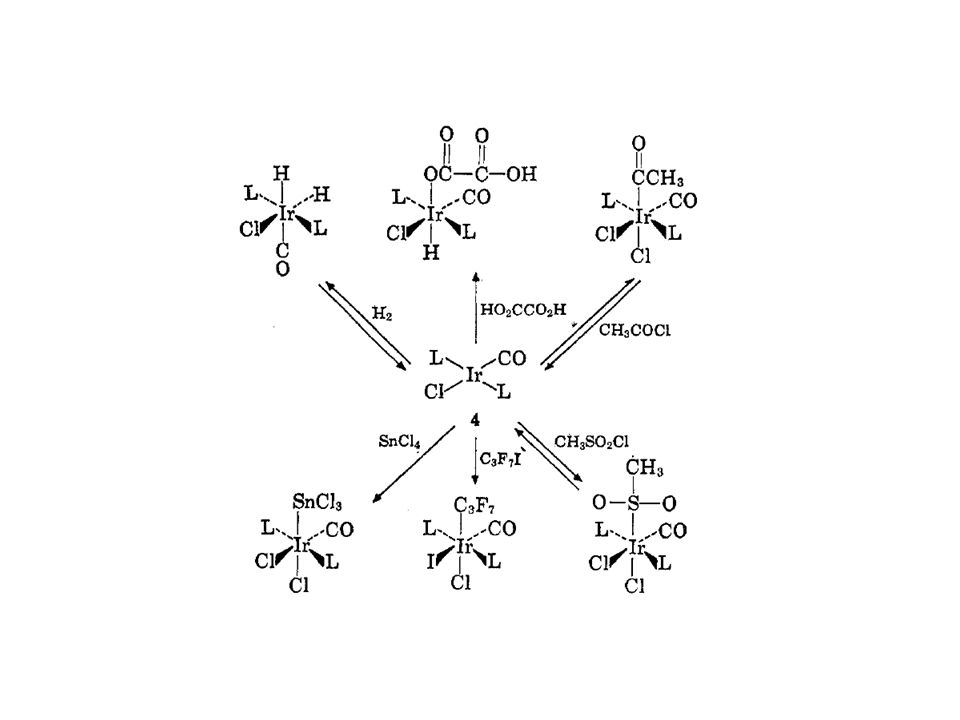
Stichwort: oxidative Addition:
Katalyse - Hydrierung oxidative Addition in Elschenbroich-Salzer: "Organometallchemie", 3. Auflage Stichwort: oxidative Addition: S. 27, 110, 172, 179, 185, 237, 240, 248, 250, 297, 481, 482, 512!

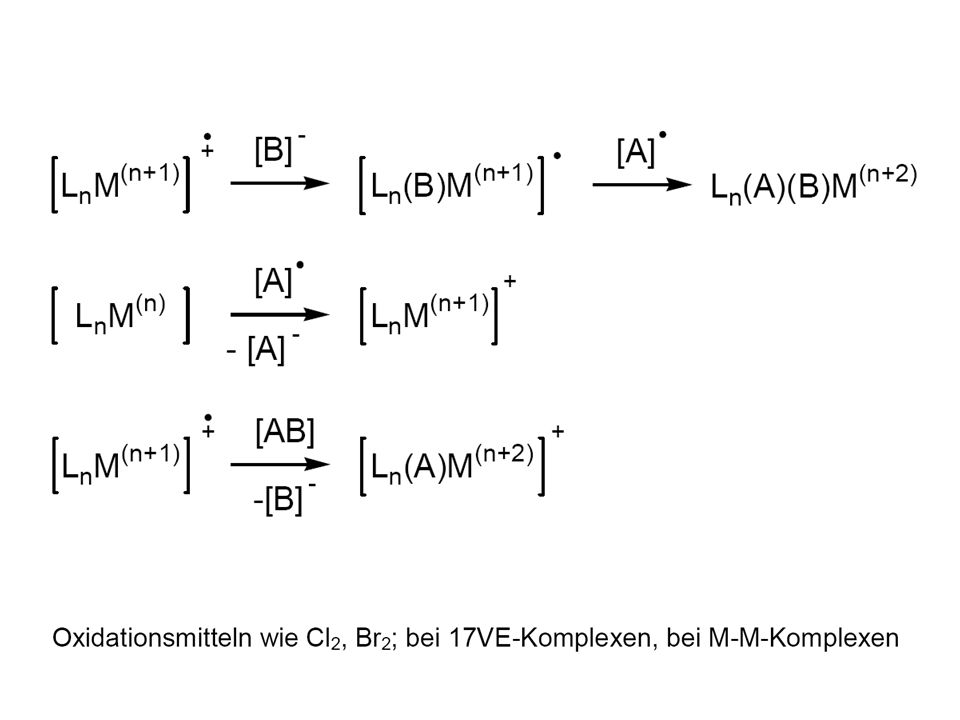
84
Oxidative Addition Reduktive Eliminierung CN: (Addition) OS: (Oxidation) # e-´s: +2
 OS: +2 (Oxidation) # e-´s: +2")
85
Wann überhaupt, für welches System einfach?
Oxidative Addition Wann überhaupt, für welches System einfach? überhaupt ? = Thermodynamik einfach ? = Kinetik # e-´s: +2 CN: (Addition) OS: (Oxidation) Parameter / "Zutaten(Gewürze)"
 OS: +2 (Oxidation) Parameter / Zutaten(Gewürze)")
86
Mechanismus ? LnM + X-Y DE Warum gefällt uns dieser Mechanismus?
konsistent mit Occam´s razor = keep it simple

87
least motion - "konzertiert" hohe Symmetrie attraktiv
DE LnM r LnM + X-Y LnM r r r least motion - "konzertiert" hohe Symmetrie attraktiv

88
least motion - "Draufschieben"/konzertiert ?
DE LnM + X-Y LnM r r vs LnM r X-Y least motion - "Draufschieben"/konzertiert ?

89
- a) b) d+ d- LnM LnM LnM..................r..............X-Y X-Y
konzertiert r LnM a) d+ d- - Orientierung: Dipol - Ladung Präferenz für b) z.B. X-Y = Med+-Id- X-Y LnM....r.... LnM r X-Y b) LnM....r....X-Y LnM....r.... X-Y konzertiert, a), oder b) ?
 d+ d- - Orientierung: Dipol - Ladung Präferenz für b) z.B. X-Y = Med+-Id- X-Y. LnM....r.... LnM r X-Y. b) LnM....r....X-Y. LnM....r.... X-Y. konzertiert, a), oder b)")
90
Alternative: LnM + X-Y LnM-X + Y• •
DG# DE LnM + X-Y a) und b) im Übergangszustand X-Y-Bdg. partiell intakt Alternative: LnM + X-Y LnM-X + Y• • 1e- Unterschied: 1e- nacheinander = Single Electron Transfer (SET)
 und b) im Übergangszustand X-Y-Bdg. partiell intakt. Alternative: LnM + X-Y LnM-X + Y• • 1e- Unterschied: 1e- nacheinander = Single Electron Transfer (SET)")
91
R-X + Mg R-Mg-X ebenfalls oxidative Addition! z.B. R-X = Me-I
Bekanntes Beispiel: Grignardreaktion R-X + Mg R-Mg-X ebenfalls oxidative Addition! z.B. R-X = Me-I ESR

92
zurück zu den Übergangsmetallen ....
später Übergangszustand da Intermediat endergon Hammond Postulat LnM + X-Y DE DG# ÜZ-produktähnlich X-Y Bindung fast komplett gebrochen DGR DG# >/ DGR LnM + X-Y LnM-X + Y• •

93
zurück zu den Übergangsmetallen ....
DG# DGR LnM + X-Y • LnM + X-Y LnM-X + Y•

94
LnM + X-Y LnM-X + Y• DG# >/ DGR DHR -{BDE(LnM-X)-BDE(X-Y)} •
z.B. CH4; BDE(C-H) = 105 kcal/mol z.B. Cp*(PMe3)Ir = BDE(Cp*(PMe3)Ir-H) = 74 kcal/mol BDE(Cp*(PMe3)Ir-Me) = 56 kcal/mol zur Erinnerung: BDE(M-Me) < BDE(M-H)
 = 105 kcal/mol. z.B. Cp*(PMe3)Ir = BDE(Cp*(PMe3)Ir-H) = 74 kcal/mol. BDE(Cp*(PMe3)Ir-Me) = 56 kcal/mol. zur Erinnerung: BDE(M-Me) < BDE(M-H)")
95
[Ir] + CH3-H [Ir]-H + CH3•
DG# >/ DGR DHR -{BDE([Ir]-H)-BDE(CH3-H)} -{74-105} = +31 kcal/mol! experimentell in sec! [Ir] + CH3-H [Ir]-CH3 + H• DG# >/ DGR DHR -{BDE([Ir]-CH3)-BDE(CH3-H)} -{56-105} = +49 kcal/mol! SET-Mechanismus kompatibel mit Experiment?
![[Ir] + CH3-H [Ir]-H + CH3•](http://slideplayer.org/slide/208366/1/images/95/%5BIr%5D+%2B+CH3-H+%EF%82%AE+%5BIr%5D-H+%2B+CH3%E2%80%A2.jpg "DG# >/ DGR DHR -{BDE([Ir]-H)-BDE(CH3-H)} -{74-105} = +31 kcal/mol! experimentell in sec! [Ir] + CH3-H [Ir]-CH3 + H• DG# >/ DGR DHR -{BDE([Ir]-CH3)-BDE(CH3-H)} -{56-105} = +49 kcal/mol! SET-Mechanismus kompatibel. mit Experiment")
96
t 109 sec 10´000d DG#30 kcal/mol RT t ® Halbwertszeiten für A
B gemäss Eyring-Gleichung 1/2 18 109 sec 10´000d 16 14 12 10 RT ) [log(sec)] 8 1y 1w 6 1d D G # =40 kcal/mol 4 1h D G # =35 kcal/mol t 1/2 2 1min DG#30 kcal/mol log( D G # =30 kcal/mol 1s D G # -2 =25 kcal/mol -4 D G # =20 kcal/mol -6 D G # =15 kcal/mol -8 D G # =10 kcal/mol -10 20 40 60 80 100 120 140 160 180 200 220 T [°C]
 [log(sec)] 8. 1y. 1w. 6. 1d. D. G. # =40 kcal/mol. 4. 1h. D. G. # =35 kcal/mol. t. 1/ min. DG#30 kcal/mol. log( D. G. # =30 kcal/mol. 1s. D. G. # -2. =25 kcal/mol. -4. D. G. # =20 kcal/mol. -6. D. G. # =15 kcal/mol. -8. D. G. # =10 kcal/mol T [°C]")
97
no way José! • [Ir] + CH3-H [Ir]-H + CH3•
DG# >/ DGR DHR -{BDE([Ir]-H)-BDE(CH3-H)} -{74-105} = +31 kcal/mol! experimentell [Ir] + CH3-H [Ir]-CH3 + H• DG# >/ DGR DHR -{BDE([Ir]-CH3)-BDE(CH3-H)} -{56-105} = +49 kcal/mol! SET-Mechanismus kompatibel mit Experiment? no way José! konzertiert oder?
![no way José! • [Ir] + CH3-H [Ir]-H + CH3•](http://slideplayer.org/slide/208366/1/images/97/no+way+Jos%C3%A9%21+%E2%80%A2+%5BIr%5D+%2B+CH3-H+%EF%82%AE+%5BIr%5D-H+%2B+CH3%E2%80%A2.jpg "DG# >/ DGR DHR -{BDE([Ir]-H)-BDE(CH3-H)} -{74-105} = +31 kcal/mol! experimentell. [Ir] + CH3-H [Ir]-CH3 + H• DG# >/ DGR DHR -{BDE([Ir]-CH3)-BDE(CH3-H)} -{56-105} = +49 kcal/mol! SET-Mechanismus kompatibel. mit Experiment no way José! konzertiert oder")
98
3 Teilchen! sehr, sehr unwahrscheinlich
für Teilchenstoß aller Wahrscheinlichkeit nach konzertiert, 3c

99
postulierter 3-Zentren Übergangszustand

100
Oxidative Addition Unterscheidung unpolare Substrate C-H, H-H, Si-H
i.d.R.mit wenigen Ausnahmen konzertierte 3-Zentren-Mechanismen Unterscheidung polare Substrate Me-I, H-Cl .. a) SN2-Typ (Substitution) b) Radikalkettenmechanismen (SET, etc.)
 SN2-Typ (Substitution) b) Radikalkettenmechanismen (SET, etc.)")
101
ox. Addition zunächst unpolare Substrate, speziell H2
wichtig z.B. für Olefin-Hydrierung ox. Addition

102
H2 MO-Vorbetrachtungen Aktivierung H2 s* E
2- Bindungsordnung = ½ {S (n e-)bindende MOs - S (n e-)antibindende MOs} = ½ { } = 1 = Einfachbindung Ox. Add.: Reduktion "H2 H22-" Bindungsordnung = ½ { } = 0! = Bindungsbruch wie werden 2 e- vom Metallzentrum transferiert?
bindende MOs - S (n e-)antibindende MOs} = ½ { } = 1 = Einfachbindung. Ox. Add.: Reduktion H2 H22- Bindungsordnung. = ½ { } = 0! = Bindungsbruch. wie werden 2 e- vom Metallzentrum. transferiert")
103
M M H2 MO-Vorbetrachtungen Aktivierung H2
Chemie Grenzorbitale = HOMO/LUMO HOMO LUMO H2 s s* E LUMO = Electrophil MLn p-Symmetrie MO´s Größe/Richtung je tieferliegend desto besser HOMO = Nucleophil M p-symm. M metall-basiert MO´s Größe/Richtung je höherliegend desto besser

104
= chemische Kreativität
MO-Vorbetrachtungen Aktivierung H2 E anhe ben = chemische Kreativität s* energet. Lage durch "Natur" festgelegt M energetisch tieferliegend!

105
H22- M H2 MO-Vorbetrachtungen Aktivierung H2 E s* M ligandbasiert s
Bindung E M s* M ligandbasiert H22-

106
M M H HOMO M H Zunahme Rückbindung/Ladungstransfer

107
symmetrieadaptiert ! ML6 Oh-Symmetrie M-L-antibindend!! nb eg* t2g eg
t1u a1g eg eg* M-L-antibindend!! symmetrieadaptiert ! t2g nb

108
b1 eg* a1 starke Absenkung hn e b2 nicht-bindend t2g d6 c4v-ML5 Oh

109
wunderbar.. p-Akzeptor H2 d6-ML5 Absenkung

110
oxidative Addition - Beispiel ML5
intakte H-H-Bindung ! R = iPr, Cyc h2-H2 Diwasserstoffkomplex LnM(H2) LnM(H)2 Evidenz???!!
 LnM(H)2. Evidenz !!")
111
zum Vergleich: n(H2) = 4395.2 cm-1
Kristallstruktur freies H2 ? H-H: 0.74 Å aufgeweitet IR (Festkörper) 0.84 Å zum Vergleich: n(H2) = cm-1 in Lösung??? Neutronenbeugung! Belege????
 0.84 Å. zum Vergleich: n(H2) = cm-1. in Lösung Neutronenbeugung! Belege")
112
NMR-Spektroskopie skalare Kopplung & Mechanismus (Fermi-Kontakt)
= g h / 4p Bo skalare Kopplung & Mechanismus (Fermi-Kontakt) H-H Abstandsbestimmung durch Messung von 1J(H-H)
 H-H Abstandsbestimmung durch Messung von 1J(H-H)")
113
NMR: Kopplung 1JHD = 43 Hz 2JHD= 0-2 Hz D
0.74 Å Å Å Å > 1.6 Å H2-Komplex "elongierter H2-Komplex" Dihydrid

114
H-D: 1JHD = 43 Hz dHH = 1.44 - 0.0168 JHD [Å] dHH dHH [Å] 1JHD [Hz]
Bindungs- ordnung 1JHD [Hz] dHH dHH = JHD [Å] empirische Formel
![H-D: 1JHD = 43 Hz dHH = JHD [Å] dHH dHH [Å] 1JHD [Hz]](http://slideplayer.org/slide/208366/1/images/114/H-D%3A+1JHD+%3D+43+Hz+dHH+%3D+JHD+%5B%C3%85%5D+dHH+dHH+%5B%C3%85%5D+1JHD+%5BHz%5D.jpg "Bindungs- ordnung. 1JHD [Hz] dHH. dHH = JHD [Å] empirische Formel.")
115
NMR-Spektroskopie = g h / 4p Bo - T1-Zeitbestimmung

117
Spin-Gitter-Relaxation => T1 Zeit Messung: Inversion Recovery
Puls (B1) z x Mxy y w1 Mo x B1 y w1 Mo z x y Relaxation z x y Spin-Gitter-Relaxation => T1 Zeit Messung: Inversion Recovery
 z. x. Mxy. y. w1. Mo. x. B1. y. w1. Mo. z. x. y. Relaxation. z. x. y. Spin-Gitter-Relaxation => T1 Zeit. Messung: Inversion Recovery.")
118
T1-Zeit Molekülbewegung & lokale Wechselfelder
Molekülbewegung (Translation/Rotation) lokale fluktuierende Wechselfelder T1-Zeit Spin-Gitter-Relaxationszeit Energieabgabe DE an Dipole des "Gitters" z.B. Lösungsmittel aber auch intramolekular!!!!!!
 lokale fluktuierende Wechselfelder. T1-Zeit Spin-Gitter-Relaxationszeit. Energieabgabe DE an Dipole des Gitters z.B. Lösungsmittel. aber auch intramolekular!!!!!!")
119
Von was hängt die T1-Zeit ab?
Molekülgröße! Korrelationszeit, tc Korrelationszeit = Zeit zwischen 2 Umorientierungen T1 ~ 1/tc

120
Von was hängt die T1-Zeit noch ab?
ebenfalls temperaturabhängig: T1 T1min T1min dominiert durch dipol. Kopplung! stark abstandsabhängig! H2-Komplexe kurze T1min < 150 msec

121
T1-Bestimmung: Inversion Recovery
z z x Mxy y p / 2 90o Detektor in x-Richtung I: maximal Imax Mo x y z x y Mo z x -Mo y I = 0 p 180o z x y Mo 3/2p 270o x -Mo y I=-Imax

124
Mz: Magnetisierung in z-Richtung
180° 90° Inversion Recovery Puls-Sequenz Mz: Magnetisierung in z-Richtung t½ = 0: Mz = - M0

128
oxidative Addition - Beispiel ML5
Oxidative Addition - Gleichgewicht? 16 e- R = iPr, Cyc LnM(H2) LnM(H)2
 LnM(H)2.")
129
R = i-Pr: Hydridbereich
im Prinzip nur Oxidation! RT 1H NMR-Spektrum R = i-Pr: Hydridbereich d ppm {31P-NMR} Verhältnis : breite Resonanzen Austausch!!!

130
verbreiterte Signale = Austausch (Lebensdauer)
gehinderte Rotation MeB MeA 1H-NMR-Spektrum, 200 MHz, RT MeC verbreiterte Signale = Austausch (Lebensdauer)
")
131
MeB MeA 1H-NMR-Spektrum, 200 MHz, RT verbreiterte Signale = Austausch Warum? Heisenberg´sche Unschärferelation: DE·Dt ~ h/2p mit DE = h·n Dn·Dt ~ 1/2p Dn ~ 1/2pDt kurze Lebendauer Dt: große Dn = breite Linien

132
K ? = k1/k-1 = 1!!! DGR = 0 DGR = 0 Austausch! aber:
DE DGR = 0 DG# groß!!! DG# klein!!! K = 1000/1000 = 1

133
Änderung? Gleichbesetzung! Signal saturiert I = 0!
Einstrahlen n(MeB) Ein strahlen n(MeB) Änderung? MeA MeB B0, E a b 16 überschüssige pro 2·106 Spins mikroskopisch DEab = gH·B0·h/2p B0, E a b makroskopisch (600 MHz) Nb/Na = e-DEab/kT = B0, E 8 Gleichbesetzung! Signal saturiert I = 0!
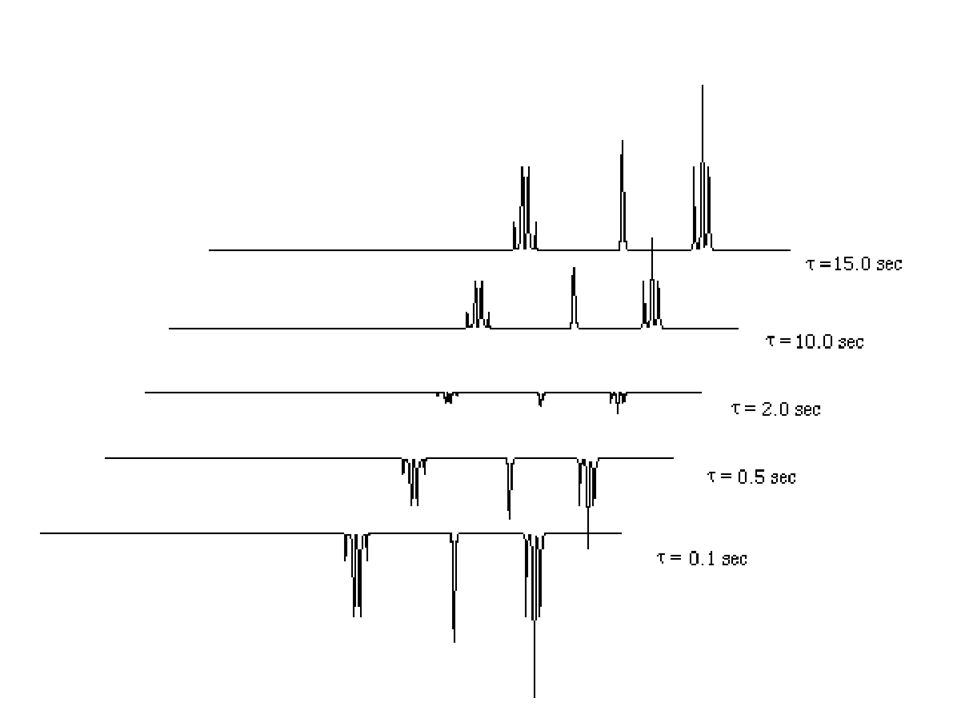
 Ein strahlen. n(MeB) Änderung MeA. MeB. B0, E. a. b. 16 überschüssige pro 2·106 Spins. mikroskopisch. DEab = gH·B0·h/2p. B0, E. a. b. makroskopisch (600 MHz) Nb/Na = e-DEab/kT = B0, E. 8. Gleichbesetzung! Signal saturiert. I = 0!")
134
I = 0 Einstrahlen n(MeB) "Spin-Saturierung" MeB MeA MeB cw-Einstr.
FID Detektionspuls I = 0 cw-Einstr. n(MeB) FID Wartezeit Warum? Intensität MeA reduziert?!! Austausch MeA MeB
 FID. Wartezeit. Warum Intensität MeA reduziert !! Austausch MeA MeB.")
135
A B

137
kobs= k1+ k-1 k-1= kobs/(1+K)
")
138
Steigung: k1+k-1 bislang unberücksichtigt Relaxation (T1) T1 schnell evtl. Korrektur
 T1 schnell evtl. Korrektur")
139
lange T1-Zeit !

141
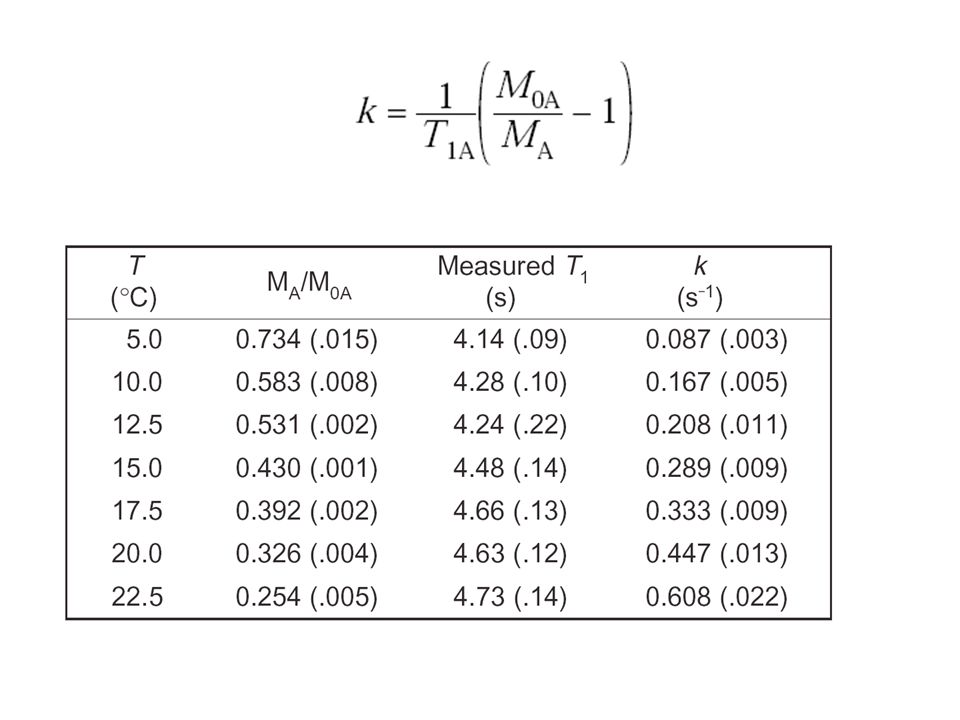
T1A T1B

143
Eyring-Darstellung DH# = 17 ± 0.4 kcal/mol DS# = 2 ± 1.0 e.u. (cal/mol·K) DG#298 18 kcal/mol schnell/langsam?

144
1) 298K: DG# 25 kcal/mol t½ 1 Tag = 86400 105 sec
Zur Erinnerung: Daumenregeln 1) 298K: DG# 25 kcal/mol t½ 1 Tag = 105 sec 2) pro 1.4 kcal/mol weniger/mehr 10x schneller/langsamer DG#298 18 kcal/mol DDG#= = 7 kcal/mol 7/1.4 = 5 => 105 x schneller t½ 105 sec/105 = 1 sec (RT)
 298K: DG# 25 kcal/mol t½ 1 Tag = 105 sec. 2) pro 1.4 kcal/mol weniger/mehr 10x schneller/langsamer. DG#298 18 kcal/mol. DDG#= = 7 kcal/mol. 7/1.4 = 5 => 105 x schneller. t½ 105 sec/105 = 1 sec (RT)")
145
Spin-Saturierungskinetik
31P-NMR Spin-Saturierungskinetik 0.04 sec-1 0.02 sec-1 1.2 DH kcal/mol Thermodynamik

146
Thermodynamik: Temperaturabhängigkeit von K
van´t Hoff Auftragung ln K(T) 1/T 1/T lnK • Achsenabschnitt: Steigung: "gutes Experiment": DT mindestens 40 K
 1/T. 1/T. lnK. • Achsenabschnitt: Steigung: gutes Experiment : DT mindestens 40 K.")
147
Verhältnis ortho/para temperaturabhängig
etwas mehr NMR-Grundlagen -PHIP Kernspin, I: z.B. 1H: I= ½, (m=+½,-½) ortho: Iges = 1, a,a, b,b, ab+ba Kernspin! para: Iges = ab - ba (Wellenfunktion) Verhältnis ortho/para temperaturabhängig RT: ortho/para = 3 : 1 80K: : 49 20K: : 998 tiefe Temperaturen para-H2 begünstigt Anreicherung
 ortho: Iges = 1, a,a, b,b, ab+ba. Kernspin! para: Iges = ab - ba (Wellenfunktion) Verhältnis ortho/para temperaturabhängig. RT: ortho/para = 3 : 1. 80K: 51 : K: 2 : 998. tiefe Temperaturen para-H2 begünstigt Anreicherung.")
148
DG# groß ortho H2 para H2 DHortho/para klein ortho/para Umwandlung langsam nutzbar für Experimente Anreicherung para-H2 (Katalyse z.B. Aktivkohle)
")
149
etwas NMR-Grundlagen Kernspin, I: z.B. 1H: I= ½, (m=+½,-½) Energie
a, m = +½ b, m = -½ Magnetfeld, Bo Zeeman-Aufspaltung NMR-Übergang (Resonanz) a,b entartet energ. günstiger g = gyromagnetisches Verhältnis
 a,b. entartet. energ. günstiger. g = gyromagnetisches Verhältnis.")
150
e- chemische Verschiebung/Abschirmung B0 B0 Beff H+ = Proton
Beff < Bo B0 e- Beff Kern H- = Hydrid H+ = Proton Abschirmung durch e-

151
e- chemische Verschiebung/Abschirmung B0 Beff H+ = Proton H- = Hydrid
Kern H- = Hydrid H+ = Proton Abschirmung durch e-

152
zwei Spin-System, AX ohne Spin-Spin-Kopplung JAX=0 aa: ab: ba: bb :

153
E bb: X ba: A ab: aa: bislang JAX=0 Berücksichtigung der Spin-Spin-Kopplung: ESS= JAX= mA·mX·h

154
ESS = JAX = mA·mX·h mit Kopplung: En,ss= En+ ESS mA = mX = +½ E1,ss= E1 + ¼·JAX·h aa mA = +½ mX = -½ E2,ss= E2 - ¼·JAX·h ab mA = -½ mX = +½ E3,ss= E3 - ¼·JAX·h ba mA = -½ mX = -½ E4,ss= E4 + ¼·JAX·h bb

155
JAX = 0 JAX > 0 E1 und E4 angehoben E bb: X1 A1 E4 ba: Spektrum
A1 A2 vA E3 X1 X2 vX A2 2 4 1 3 1 2 3 4 ab: E2 X2 aa: E1 JAX = 0

156
JAX = 0 JAX < 0 E1 und E4 abgesenkt E bb: E4 X2 A2 ba: Spektrum
A1 A2 vA A1 JAX /4 E3 X1 X2 vX 1 3 2 4 3 1 2 ab: X1 JAX /4 E2 aa: E1 JAX /4 JAX = 0 JAX < 0 E1 und E4 abgesenkt

157
E bb 4 ba 3 ab 2 aa 1

158
para: Iges = ab - ba populiert
4 ba 3 Besetzungs- änderung ab 2 aa 1

159
Intensitätssteigerung!!
para: Iges = ab - ba populiert E bb 4 Intensitätssteigerung!! ba 3 Emission!! ab 2 aa 1

160
1H-NMR-Spektrum nach 40 sec bei 48°C
PHIP 20 H´s 2 H´s !

161
Deutung? DG#transCl DG#transCO DDG# DDGR nach längerer Zeit
kinetisches Produkt entsteht schneller DG#transCl DDG# DG#transCO thermodyn. Produkt energet. günstiger DDGR Deutung?

162
DDG# = 1.4 kcal/mol = 10x schneller 2.8 kcal/mol = 100x "
[IrtransCO] [IrtransCl] = ktransCO ktransCl DDG# = 1.4 kcal/mol = 10x schneller 2.8 kcal/mol = 100x " k

163
DG#transCl DG#transCO DDG# DDGR nach längerer Zeit kinetisches Produkt
entsteht schneller DG#transCl DDG# DG#transCO thermodyn. Produkt energet. günstiger DDGR

164
Thermodynamik DDGR [kcal/mol]
![Thermodynamik DDGR [kcal/mol]](http://slideplayer.org/slide/208366/1/images/164/Thermodynamik+DDGR+%5Bkcal%2Fmol%5D.jpg "Thermodynamik DDGR [kcal/mol]")
165
Oxidative Addition von C-H-Bindungen = C-H-Aktivierung

166
isolobal: Grenzorbitale: gleiche Symmetrie
oder isolobal: Grenzorbitale: gleiche Symmetrie & Ausdehnung „vergleichbare Chemie“

167
Oxidative Addition von C-H-Bindungen = C-H-Aktivierung

168
Ir Kinetik ? bimolekular "1.0000" ÜZ

169
Überprüfung der Reaktionsordnung - Bed. pseudoerster Ordnung
1 Komponente im großen Überschuß mindestens > 10-15x konstant! z.B.: vor Reaktionsbeginn: 100 eq. C6H6 nach Reaktionsende: eq. C6H6 Änderung: /100 = 1 % 0 %!

170
Geradengleichung kobs t Steigung: kobs experimentell beobachtet!

171
kobs = k·[C6H6] [C6H6] Überprüfung Reaktion 2. Ordnung
Messung von kobs bei verschiedenen [C6H6] [C6H6] Steigung: k erwartet Gerade
![kobs = k·[C6H6] [C6H6] Überprüfung Reaktion 2. Ordnung](http://slideplayer.org/slide/208366/1/images/171/kobs+%3D+k%C2%B7%5BC6H6%5D+%5BC6H6%5D+%C3%9Cberpr%C3%BCfung+Reaktion+2.+Ordnung.jpg "Messung von kobs bei verschiedenen [C6H6] [C6H6] Steigung: k. erwartet Gerade.")
172
Erwartung Experiment kobs [C6H6] [C6H6] Sättigung Deutung ?
Steigung: k Erwartung Experiment [C6H6] kobs Sättigung Deutung ?
![Erwartung Experiment kobs [C6H6] [C6H6] Sättigung Deutung](http://slideplayer.org/slide/208366/1/images/172/Erwartung+Experiment+kobs+%5BC6H6%5D+%5BC6H6%5D+S%C3%A4ttigung+Deutung.jpg "Steigung: k. Erwartung. Experiment. [C6H6] kobs. Sättigung. Deutung")
173
= 0!! Oxidative Addition von C-H-Bindungen = C-H-Aktivierung
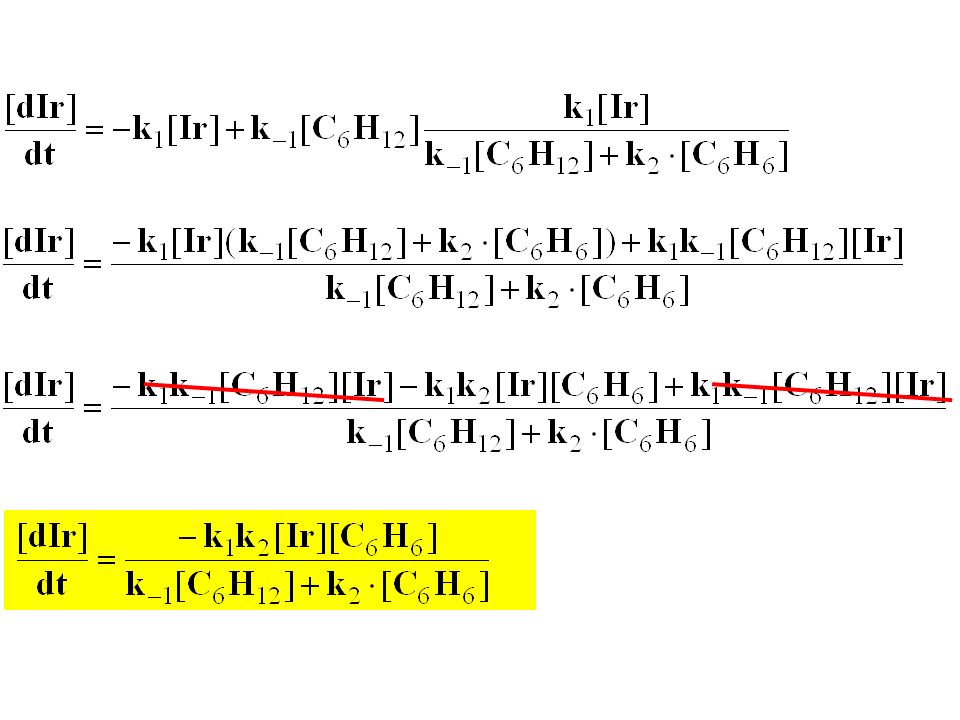
Ir Ir2 k1 k-1 k2 Mikroskopische Reversibilität! Kinetik? = 0!! Bodenstein (quasi-stationär)
")
174
(1) in (1) einsetzen Hausaufgabe
 in (1) einsetzen Hausaufgabe")
176
[C6H6] klein: k2[C6H6] << k-1[C6H12]
![[C6H6] klein: k2[C6H6] << k-1[C6H12]](http://slideplayer.org/slide/208366/1/images/176/%5BC6H6%5D+klein%3A+k2%5BC6H6%5D+%3C%3C+k-1%5BC6H12%5D.jpg "[C6H6] klein: k2[C6H6] << k-1[C6H12]")
177
Gerade unabhängig von [C6H6]
[C6H6] groß: k2[C6H6] >> k-1[C6H12] Gerade unabhängig von [C6H6]
![Gerade unabhängig von [C6H6]](http://slideplayer.org/slide/208366/1/images/177/Gerade+unabh%C3%A4ngig+von+%5BC6H6%5D.jpg "[C6H6] groß: k2[C6H6] >> k-1[C6H12] Gerade unabhängig von [C6H6]")
178
[C6H6] kobs Sättigung
![[C6H6] kobs Sättigung](http://slideplayer.org/slide/208366/1/images/178/%5BC6H6%5D+kobs+S%C3%A4ttigung.jpg "[C6H6] kobs Sättigung")
179
Einfluß? kinetischer Isotopeneffekt

180
etwas Schwingungsspektroskopie

181
Morsepotential A-B A· + B· A· + B· A-B Dissozation senergie
E = (1+½)hn E = (0+½)hn (Nullpunktsschwingung)
hn. E = (0+½)hn (Nullpunktsschwingung)")
182
harmonischer Oszillator
A-B A· + B· A· + B· A-B n ~ k/m n: Schwingungsfrequenz k: Kraftkonstante m: reduzierte Masse m = mA·mB /mA+ mB DE = hn IR

183
harmonischer Oszillator
R3C-D R3C· + D· R3C-H R3C· + H· R3C-H R3C· + H· R3C· + D· R3C-D n ~ k/m kC-D kC-H aber m: mCD = 12·2/12+2=24/ mCH = 12·1/12+1=12/13 R3C-H R3C-D nC-H/nC-D ~ k/mCH / k/mCH = 13/12 / 14/24 2 = 13·24/12·14

184
harmonischer Oszillator
R3C-D R3C· + D· R3C-H R3C· + H· R3C-H R3C· + H· R3C· + D· R3C-D Dissozations enthalpie C-D C-H R3C-H DDG R3C-D

185
DGD# - DGH# = ½ hnR-H - ½ hnR-D
Übergangszustand ÜZ DG#H DG#D DGD# - DGH# = ½ hnR-H - ½ hnR-D DGD# - DGH# R-H R-D Eyring: kH,D = kBT/h · e-DGH,D#/RT kH / kD = e-DGH#/RT / e-DGD#/RT kH / kD = e-(DGH#-DGD#)/RT = e(DGD#-DGH#)/RT kH / kD = e(nR-H - nR-H)·½h/kT
/RT = e(DGD#-DGH#)/RT. kH / kD = e(nR-H - nR-H)·½h/kT.")
186
kH / kD = enR-H·7.069·10-4 kH / kD = e(nR-H - nR-D)·h/2kT
mit nH/nD 2 kH / kD = e(nR-H - nR-H/2 )·h/2kT = enR-H·(1-½2 )·h/2kT bei RT: nR-H in [cm-1] kH / kD = enR-H·7.069·10-4
·h/2kT = enR-H·(1-½2 )·h/2kT. bei RT: nR-H in [cm-1] kH / kD = enR-H·7.069·10-4.")
187
Maximale Isotopeneffekte
kH / kD = enR-H·7.069·10-4 n(O-H) n(C-H) 8 bei RT! n(S-H) n(M-H)
 n(C-H) 8. bei RT! n(S-H) n(M-H)")
188
Maximale Isotopeneffekte
T = 25 °C T = 100 °C T = -50 °C stark temperaturabhängig

189
RT: kC-H /C-D = 40 Maximale Isotopeneffekte quantenmech.Tunneln
Welle/Teilchen Dualismus stark massenabhängig H (m=1) tunnelt("wellt"), D (m=2) tunnelt nicht allerdings temperaturunabhängig!
 tunnelt( wellt ), D (m=2) tunnelt nicht. allerdings temperaturunabhängig!")
190
andere Isotope k k Isotop leicht schwer C-H/C-D 6 - 8 C-H/C-T 15 - 16
(bei 25 C ) schwer C-H/C-D 6 - 8 C-H/C-T 12 13 C / C 1.04 12 14 C / C 1.07 14 15 N/ N 1.03 16 18 O / O 1.02 32 34 S/ S 1.01 35 37 Cl/ Cl 1.01
 schwer. C-H/C-D C-H/C-T C. / C C. / C N/ N O. / O S/ S Cl/ Cl")
191
primärer Isotopeneffekt: Isotop direkt an Bindungsbruch beteiligt
kH/kD ? = 6.7 primärer Isotopeneffekt: Isotop direkt an Bindungsbruch beteiligt Deutung? E2-Mechanismus C-H,D Bdgs.-Bruch im ÜZ

192
primärer Isotopeneffekt
kH/kD ? = 1.4 Deutung? E1-Mechanismus - H+, D+ C-Br Bdgs.-Bruch im ÜZ

193
electrophile aromatische Substitution
kH/kD ? = 1.0 electrophile aromatische Substitution Wheland-Zwischenprodukt

194
sekundäre Isotopeneffekte
H3C-Cl + H2O H3C-OH + HCl kH D3C-Cl + H2O D3C-OH + HCl kD Exp.: kH /kD = 0.97

195
CH I - I k Substrat k Exp: 1.001 1.009 1.030 1.095 N N CH 3 3 D H CD 3
+ 3 N N - + I CH 3 k Substrat k Exp: D / H CD 3 1.001 N CD 3 1.009 N 1.030 N CD 3 1.095 D C N CD 3 3

196
Hyperkonjugativer Effekt
(CH3)C-Cl + H2O (CH3)C-OH + HCl kH (CD3)C-Cl + H2O (CD3)C-OH + HCl kD Exp.: kH /kD = 1.21 sterischer Effekt
C-Cl + H2O (CH3)C-OH + HCl kH. (CD3)C-Cl + H2O (CD3)C-OH + HCl kD. Exp.: kH /kD = sterischer Effekt.")
197
sekundärer Istopeneffekt

198
Gleichgewichtsisotopeneffekt I - sekundär
+ z.B. CH COOH CH COO + H K 3 3 H - + CD COOH CD COO + H K 3 3 D exp.: K / K = 1.06 H D Begründung: Wasserstoff etwas mehr elektronegativ als Deuterium D-: näher an X: besserer Donor

199
Gleichgewichtsisotopeneffekt I primär A-H-Bindung stärker als O-H
A-H + H2O A- + H3O+ pKH pKD A-D + H2O A- + D3O+ exp.: pKD - pKH = 0.6 AH saurer! Deutung ? A-H-Bindung stärker als O-H

200
Das schwerere Isotop bevorzugt die stärkere Bindung!!
H,D3O+ A-H,D stärkere Bindung größere Nullpunkts- schwingungdifferenz Das schwerere Isotop bevorzugt die stärkere Bindung!!

201
zu "invers" für reine red. Eliminierung (rein kin. IE)
kH/kD = 0.7 zu "invers" für reine red. Eliminierung (rein kin. IE)
")
202
Gleichgewichtsisotopeneffekt
n(M-H) 2000 cm-1 n(C-H) 3000 cm-1 IR:
 2000 cm-1. n(C-H) 3000 cm-1. IR:")
203
intramolekularer H,D-Austausch
Weitere Hinweise H D intramolekularer H,D-Austausch

204
Gleichgewichtsisotopeneffekt endotherm (exp. bestätigt)
produktähnlich Hammond-Postulat

205
kinetischer Isotopeneffekt kinetischer Isotopeneffekt
inverser primärer kinetischer Isotopeneffekt normaler primärer kinetischer Isotopeneffekt normaler primärer KIE

206
S

207
x# Hammond-Postulat 1 EEdukt = k·x2 EProdukt = k·(x-1)2 + DE0
EEdukt = k·x2 EProdukt = k·(x-1)2 + DE0 ÜZ: EEdukt = EProdukt k·x#2 = k·(x#-1)2 + DE0 x# = ½ +DE0/2k DE#=E(x#)=k(½ +DE0/2k)2 = k/4 + DE0/2 + DE02/4k
2 + DE0. ÜZ: EEdukt = EProdukt. k·x#2 = k·(x#-1)2 + DE0. x# = ½ +DE0/2k. DE#=E(x#)=k(½ +DE0/2k)2 = k/4 + DE0/2 + DE02/4k.")
208
x# 1 x# = ½ +DE0/2k DE0= 0 x# = ½ DE0< 0 x# < ½
DE# x# E Edukt DE0 1 Produkt x# = ½ +DE0/2k DE0= x# = ½ "ÜZ mittig" DE0< x# < ½ früher ÜZ näher an Edukt DE0> x# > ½ später ÜZ näher an Produkt

209
intrinsische Barriere DEo# für DE0=0 (thermoneutral)
DE0=0: DEo# = k/4 + DE0/2 + DE02/4k k = 4DE0# x# = ½ +DE0/8DE0# Marcus Theorie DE# = DE0# + DE0/2 + DE02/16DE0# quadratischer Term in der Regel vernachlässigbar! (außer für DE0 >> 0 oder DE0 <<0) DE# = DE0# + DE0/2
 DE# = DE0# + DE0/2.")
210
DE#= DE0# + DE0/2 + DE02/16DE0# Minimum! exothermer

211
Si-H Aktivierung Bedeutung für Hydrosilylierung:

212
Homolytische Aktivierung

213
Abstoßung => monomer!
N4Rh· N4Rh-N4Rh Dimer Metall-Metall-Bindung Gleichgewicht dxz q.planar: dx2-y2 dz2 dxy dyz

214
2 RT! + R-H R = Me, C6H5 Kinetik: v = kobs·[N4Rh]2·[R-H]
∆H# = 7.1 kcal/mol ∆S# = -39 eu (R=Me) ∆G#(298 K) = ∆H# -T∆S# = ·(-39) = 19 kcal/mol 3. Ordnung! kin. Isotopeneffekt kH/kD = 8 (maximal)!
![2 RT! + R-H R = Me, C6H5 Kinetik: v = kobs·[N4Rh]2·[R-H]](http://slideplayer.org/slide/208366/1/images/214/2+RT%21+%2B+R-H+R+%3D+Me%2C+C6H5+Kinetik%3A+v+%3D+kobs%C2%B7%5BN4Rh%5D2%C2%B7%5BR-H%5D.jpg "∆H# = 7.1 kcal/mol ∆S# = -39 eu (R=Me) ∆G#(298 K) = ∆H# -T∆S# = ·(-39) = 19 kcal/mol. 3. Ordnung! kin. Isotopeneffekt kH/kD = 8 (maximal)!")
215
t t1/2 1 min! DG#19 kcal/mol 298K t ® Halbwertszeiten für A
B gemäss Eyring-Gleichung log( t 1/2 ) [log(sec)] 1/2 18 16 14 12 10 8 1y 1w 6 1d D G # =40 kcal/mol 4 1h D G # =35 kcal/mol 2 t1/2 1 min! 1min 298K D G # =30 kcal/mol 1s # -2 D G =25 kcal/mol DG#19 kcal/mol -4 D G # =20 kcal/mol -6 D G # =15 kcal/mol -8 D G # =10 kcal/mol -10 20 40 60 80 100 120 140 160 180 200 220 T [°C]
 [log(sec)] 1/ y. 1w. 6. 1d. D. G. # =40 kcal/mol. 4. 1h. D. G. # =35 kcal/mol. 2. t1/2 1 min! 1min. 298K. D. G. # =30 kcal/mol. 1s. # -2. D. G. =25 kcal/mol. DG#19 kcal/mol. -4. D. G. # =20 kcal/mol. -6. D. G. # =15 kcal/mol. -8. D. G. # =10 kcal/mol T [°C]")
216
2 RT! + R-H R = Me, C6H5 Kinetik: v = kobs·[N4Rh]2·[R-H]
3. Ordnung! kin. Isotopeneffekt kH/kD = 8 (maximal)! ∆H# = 7.1 kcal/mol ∆S# = -39 eu (R=Me) ∆G#(298 K) = ∆H# -T∆S# = ·(-39) = 19 kcal/mol
![2 RT! + R-H R = Me, C6H5 Kinetik: v = kobs·[N4Rh]2·[R-H]](http://slideplayer.org/slide/208366/1/images/216/2+RT%21+%2B+R-H+R+%3D+Me%2C+C6H5+Kinetik%3A+v+%3D+kobs%C2%B7%5BN4Rh%5D2%C2%B7%5BR-H%5D.jpg "3. Ordnung! kin. Isotopeneffekt kH/kD = 8 (maximal)! ∆H# = 7.1 kcal/mol ∆S# = -39 eu (R=Me) ∆G#(298 K) = ∆H# -T∆S# = ·(-39) = 19 kcal/mol.")
217
Postulierter Übergangszustand
Verbesserung?! intramolekular

218
X LnM R X Polare Substrate HX: HCl
R-X: Methyliodid, Phenyliodid, Methyltriflat.... in der Regel: keine konzertierten 3-Zentren-Mechanismen X LnM R X

219
= Industrielle Essigsäuredarstellung - Homogene Katalyse O
Carbonylierung von Methanol CH3-OH + CO ® CH3-C-OH = O 7 Millionen Jahrestonnen - bis 1960 Fa. BASF Cobalt-basierend 200°C, 700 bar, geringe Selektivität - ab 1970 Fa. Monsanto Rhodium-basierend 150°C, 200 bar, Monsanto-Prozess Fa. BP übernimmt „Monsanto-Prozess“ ab 1996 Fa. BP „Cativa Prozess“ Iridium-basierend höhere Selektivität

220
Katalyse Übersicht "Organik" -d[CH3OH] -dt = k [Rh] ● [CH3I]
![Katalyse Übersicht Organik -d[CH3OH] -dt = k [Rh] ● [CH3I]](http://slideplayer.org/slide/208366/1/images/220/Katalyse+%C3%9Cbersicht+Organik+-d%5BCH3OH%5D+-dt+%3D+k+%5BRh%5D+%E2%97%8F+%5BCH3I%5D.jpg "Katalyse Übersicht Organik -d[CH3OH] -dt = k [Rh] ● [CH3I]")
221
? cis oder trans ? Vaska´s Komplex Ir(I), d8-konfig. q.pl.
"Drosophila" Ir(I), d8-konfig. q.pl. polare LM: (DMF, MeOH, H2O, MeCN) cis + trans unpolare LM: (C6H6, CHCl3) nur cis Gasphase nur cis
, d8-konfig. q.pl. polare LM: (DMF, MeOH, H2O, MeCN) cis + trans unpolare LM: (C6H6, CHCl3) nur cis Gasphase nur cis.")
222
Collman´s Reagenz

224
parallel perpendicular

225
Thermodynamik + R-I DH R-I DH [kJ/mol]!! stark exotherm
![Thermodynamik + R-I DH R-I DH [kJ/mol]!! stark exotherm](http://slideplayer.org/slide/208366/1/images/225/Thermodynamik+%2B+R-I+DH+R-I+DH+%5BkJ%2Fmol%5D%21%21+stark+exotherm.jpg "Thermodynamik + R-I DH R-I DH [kJ/mol]!! stark exotherm")
226
Kinetik & DS# << 0 v = kobs·[Ir][Y-Z] Reaktion 2. Ordnung *
X Y-Z k [M-1sec-1] DH# [kcal/mol] DS# [eu] *30°C, Benzol *
![Kinetik & DS# << 0 v = kobs·[Ir][Y-Z] Reaktion 2. Ordnung *](http://slideplayer.org/slide/208366/1/images/226/Kinetik+%26+DS%23+%3C%3C+0+v+%3D+kobs%C2%B7%5BIr%5D%5BY-Z%5D+Reaktion+2.+Ordnung+%2A.jpg "X Y-Z k [M-1sec-1] DH# [kcal/mol] DS# [eu] *30°C, Benzol. *")
227
l a n g s m e r Deutung? Sterik, Elektronik

228
Sicht von oben auf Ebene Sicht in Ebene Angriff

229
Sterik Tolman Kegelwinkel Systematisierung

230
l a n g s m e r Deutung? Sterik, Elektronik

231
l a n g s m e r besserer Donor

232
Elektronische Parameter
Systematik Tolman - Parameter basierend auf n(CO)
")
233
zusätzliche Beobachtungen
Zusammenfassung bis jetzt - 2. Ordnung - DS# << 0 - schneller für e--reiche Komplexe - Stereochemie - k(Ox. Add.): R-X: X = OTf > I > Tos Br > Cl - beschleunigt in polaren LM zusätzliche Beobachtungen
: R-X: X = OTf > I > Tos Br > Cl. - beschleunigt in polaren LM. zusätzliche Beobachtungen.")
234
weiterer Hinweis - isolierbares Zwischenprodukt

235
Deutung

236
Mechanismus Me-I Nucleophil Electrophil LUMO s* HOMO d+ d- dxz
q.planar: dx2-y2 dz2 dxy dyz HOMO LUMO s*

237
Nucleophil

238
tbp + + I- I- trans-Konfiguration Nucleophil
oktaedrisch d6-konfiguriert

240
tbp q.-py. E d6-Konfiguration q.py. bevorzugt d6-Konfiguration = 180°

241
"Wie heißt das Kind?" SN2-Mechanismus
Nucleophil tbp + I- + I- "Wie heißt das Kind?" SN2-Mechanismus

242
stereoselektiv: Inversion
SN2-Mechanismus ? Stereochemie ? Nu| * + Y| stereoselektiv: Inversion Walden-Umkehr

243
Stereochemie - Experiment Inversion!

244
Beschleunigung durch I- Zugabe!

245
Oxidative Addition - geschwindigkeitsbestimmend
ersichtlich aus Geschwindigkeitsgesetz: -d[CH3OH] -dt = k [Rh] ● [CH3I] Erhöhung der Raum-Zeit-Ausbeute $,$,$,$ !!!

246
Deutung (Monsanto-System) at-Komplex-Bildung besseres Nucleophil!
 at-Komplex-Bildung besseres Nucleophil!")
247
groß durch pzZumischung guter Überlapp (S groß)
= LUMO Electrophil guter Überlapp (S groß) DE ~ DS2/Ei-Ej
 DE ~ DS2/Ei-Ej.")
248
Früheres Experiment zur Stereochemie
funktioniert reproduzierbar nur in Anwesenheit von O2

249
Oxidative Addition - Radikalmechanismus
Solvenskäfig +

250
SET:

252
Br - PhCH2· Br PhCH2Br 2 PhCH2· PhCH2-CH2Ph
+ Dissoziation aus Käfig Br - PhCH2· Br PhCH2Br 2 PhCH2· PhCH2-CH2Ph

253
LnM LnM + + 2e--Mechanismus "normale Prod."

254
Belege LnM LnM + + • SET - Br- +

255
"radical clocks" • • k(298K)=2.1·108 sec-1 t1/2 = 3.3 10-9 sec!
"pfeilschnell"

256
Zeitskala Eyring: k = kB/T·h·e-DG#/RT
Annahme: DG# = 0: k = kB·T/h·e0 = kB/T·h bei 298 K: k = 1.38·10-23·298/6.6·10-34 = 6.2·1012 sec-1 schnellstmögliche intramolekulare Geschwindigkeitskonstante Oxidative Addition: aber intermolekular 20 Å keine Reaktion diffusionskontrolliert! Diffusion

257
Zeitskala Eyring: k = kB/T·h·e-DG#/RT
Annahme: DG# = 0: k = kB·T/h·e0 = kB/T·h bei 298 K: k = 1.38·10-23·298/6.6·10-34 = 6.2·1012 sec-1 schnellstmögliche intramolekulare Geschwindigkeitskonstante Oxidative Addition: aber intermolekular diffusionskontrolliert! 3 Å Reaktion! Diffusion: langsam!

258
bimolekulare Reaktion!
Diffusionskontrolle Diffusion: langsam! 20 Å keine Reaktion Es gilt: <x> = 2·D·t t = Zeit D = Diffusionskonstante <x> = = 17Å <x> = 17·10-10 m t = <x>2/2·D 3 Å Reaktion Dtypisch = 10-9 m2/sec t = (17·10-10)2/2·10-9 = sec NB: <x> = 1 cm t = sec 1/2 Tag!!!!! !!Rühren!! (Konvektion) kmax = 1/t 109 l/mol·sec Obergrenze: bimolekulare Reaktion!
2/2·10-9 = sec. NB: <x> = 1 cm. t = sec 1/2 Tag!!!!! !!Rühren!! (Konvektion) kmax = 1/t 109 l/mol·sec. Obergrenze: bimolekulare Reaktion!")
259
Mischung: radikalisch + SN2
• "radical clocks" • k(298K)=2.1·108 sec-1 kmax = 1/t 109 l/mol·sec Wenn ausschließlich SN2 Mischung: radikalisch + SN2 zu "99.99 %"
=2.1·108 sec-1. kmax = 1/t 109 l/mol·sec. Wenn. ausschließlich. SN2. Mischung: radikalisch + SN2. zu %")
260
Experiment

Ähnliche Präsentationen





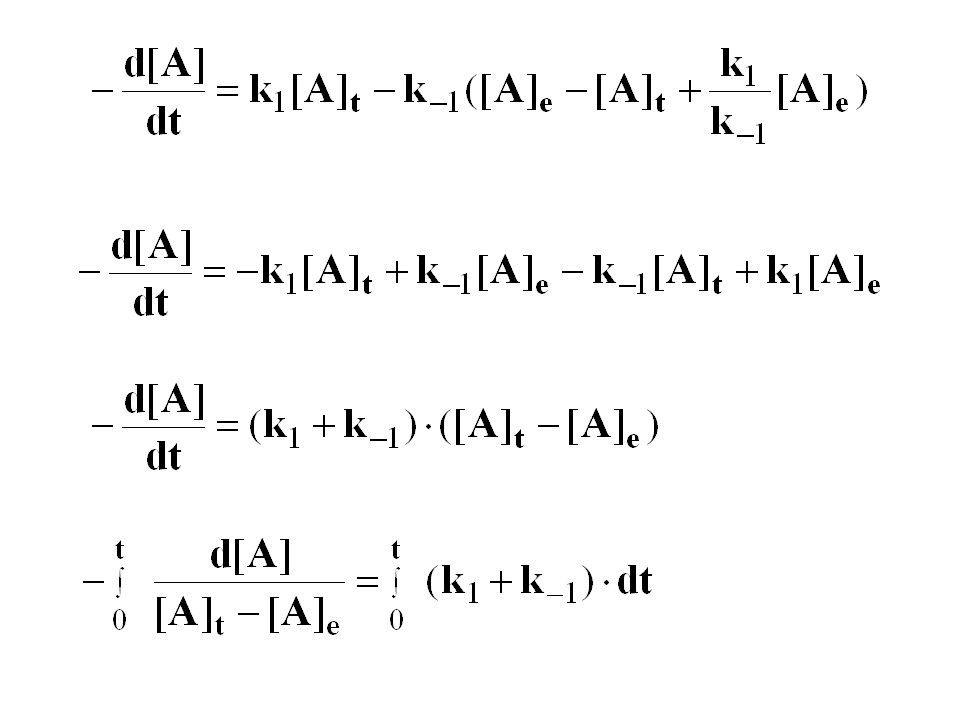



>")








>")
>")
>")
>")




